ChIP-exo
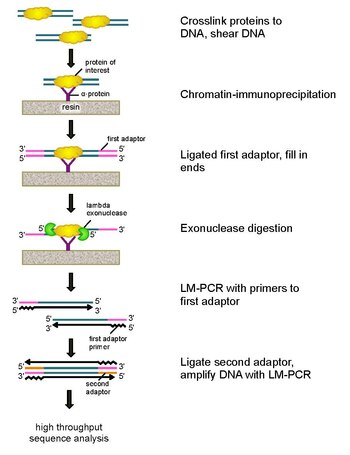
ChIP-exo to oparta na immunoprecypitacji chromatyny metoda mapowania miejsc, w których białko będące przedmiotem zainteresowania ( czynnik transkrypcyjny ) wiąże się z genomem. Jest to modyfikacja ChIP-seq , poprawiająca rozdzielczość miejsc wiązania z setek par zasad do prawie jednej pary zasad. Wykorzystuje użycie egzonukleaz do degradacji nici DNA związanego z białkiem w kierunku 5'-3' do niewielkiej liczby nukleotydów miejsca wiązania białka. Nukleotydy końców traktowanych egzonukleazą są określane przy użyciu pewnej kombinacji sekwencjonowania DNA , mikromacierzy i PCR . Sekwencje te są następnie mapowane do genomu, aby zidentyfikować miejsca w genomie, w których wiąże się białko.
Teoria
immunoprecypitacji chromatyny ( ChIP ) są stosowane od 1984 roku do wykrywania interakcji białko-DNA. Istnieje wiele odmian ChIP w celu poprawy jakości wyników. Jedno z takich ulepszeń, ChIP-on-chip (ChIP-chip), łączy ChIP z technologią mikromacierzy. Ta technika ma ograniczoną czułość i specyficzność, zwłaszcza in vivo , gdzie mikromacierze są ograniczone przez tysiące białek obecnych w kompartmencie jądrowym, co skutkuje wysokim odsetkiem wyników fałszywie dodatnich. Następnie pojawiło się sekwencjonowanie ChIP (ChIP-seq), które łączy ChIP z sekwencjonowaniem o dużej przepustowości. Jednak heterogeniczny charakter ściętych fragmentów DNA odwzorowuje miejsca wiązania z dokładnością do ±300 par zasad, co ogranicza specyficzność. Po drugie, zanieczyszczenie DNA stanowi poważny problem, ponieważ tak niewiele loci genetycznych jest usieciowanych z białkiem będącym przedmiotem zainteresowania, co sprawia, że każdy niespecyficzny genomowy DNA jest znaczącym źródłem szumu tła.
Aby rozwiązać te problemy, Rhee i Pugh zrewidowali klasyczny test ochrony przed nukleazami , aby opracować ChIP-exo. Ta nowa technika ChIP opiera się na egzonukleazie lambda , która degraduje tylko i wszystkie niezwiązane dwuniciowe DNA w kierunku 5′-3′. Pokrótce, białko będące przedmiotem zainteresowania (konstruowanie jednego ze znacznikiem epitopowym może być przydatne do immunoprecypitacji) jest sieciowane in vivo z jego naturalnymi miejscami wiązania w całym genomie przy użyciu formaldehydu. Komórki są następnie zbierane, rozbijane, a chromatyna jest ścinana i solubilizowana przez sonikację . Następnie do immunoprecypitacji białka będącego przedmiotem zainteresowania stosuje się przeciwciało wraz z usieciowanym DNA. Adaptery DNA PCR są następnie ligowane z końcami, które służą jako punkt startowy do syntezy drugiej nici DNA po trawieniu egzonukleazą. Następnie egzonukleaza lambda trawi podwójne nici DNA od końca 5', aż do zablokowania trawienia na granicy kowalencyjnego oddziaływania białko-DNA. Większość zanieczyszczającego DNA jest degradowana przez dodanie drugiej egzonukleazy specyficznej dla pojedynczej nici. Po sieciowania startery do adapterów PCR są wydłużane w celu utworzenia dwuniciowego DNA, a drugi adapter jest ligowany z końcami 5 'w celu dokładnego wyznaczenia miejsca zakończenia trawienia egzonukleazą. Bibliotekę następnie amplifikuje się metodą PCR, a produkty identyfikuje się za pomocą wysokowydajnego sekwencjonowania . Ta metoda pozwala na rozdzielczość do pojedynczej pary zasad dla dowolnego miejsca wiązania białka w dowolnym genomie, co jest znacznie wyższą rozdzielczością niż w przypadku ChIP-chip lub ChIP-seq.
Zalety
Wykazano, że ChIP-exo poddaje się rozdzielczości pojedynczej pary zasad w identyfikowaniu miejsc wiązania białek. Jest to w przeciwieństwie do ChIP-seq, który może zlokalizować miejsce wiązania białka tylko z ± 300 par zasad.
Zanieczyszczenie fragmentów DNA niezwiązanych z białkami może skutkować wysokim odsetkiem wyników fałszywie dodatnich i ujemnych w eksperymentach ChIP. Dodanie egzonukleaz do procesu nie tylko poprawia rozdzielczość wywoływania miejsc wiązania, ale także usuwa zanieczyszczający DNA z roztworu przed sekwencjonowaniem.
Białka, które są nieefektywnie związane z fragmentem nukleotydu, są bardziej podatne na wykrycie przez ChIP-exo. Umożliwiło to na przykład rozpoznanie większej liczby miejsc wiązania czynnika transkrypcyjnego CTCF niż wcześniej odkryto.
Ze względu na wyższą rozdzielczość i zmniejszone tło, podczas korzystania z ChIP-exo wymagana jest mniejsza głębokość pokrycia sekwencjonowaniem.
Ograniczenia
Jeśli kompleks białko-DNA ma wiele miejsc usieciowania w ramach pojedynczego zdarzenia wiązania, może wyglądać tak, jakby istniało wiele różnych zdarzeń wiązania. Prawdopodobnie wynika to z denaturacji i sieciowania tych białek w jednym z dostępnych miejsc wiązania w ramach tego samego zdarzenia. Egzonukleaza zatrzymałaby się wówczas w jednym z miejsc związanych, w zależności od miejsca, w którym białko jest usieciowane.
Podobnie jak w przypadku każdej metody opartej na ChIP, odpowiednie przeciwciało dla białka będącego przedmiotem zainteresowania musi być dostępne, aby można było zastosować tę technikę.
Aplikacje
Rhee i Pugh wprowadzają ChIP-exo, przeprowadzając analizy na niewielkim zbiorze czynników transkrypcyjnych: Reb1, Gal4, Phd1, Rap1 u drożdży i CTCF u człowieka. Miejsca Reb1 często znajdowano w klastrach, a te klastry miały około 10-krotnie większe obłożenie niż oczekiwano. Wtórne miejsca w klastrach znaleziono ~40 pz od głównego miejsca wiązania. Motywy wiążące Gal4 wykazywały silną preferencję dla trzech z czterech nukleotydów, co sugeruje negatywną interakcję między Gal4 a wykluczonym nukleotydem. Phd1 rozpoznaje trzy różne motywy, co wyjaśnia wcześniejsze doniesienia o niejednoznaczności motywu wiążącego Phd1. Stwierdzono, że Rap1 rozpoznaje cztery motywy. Geny białek rybosomalnych związane z tym białkiem miały tendencję do wykorzystywania określonego motywu o silniejszej sekwencji konsensusowej. Inne geny często wykorzystywały skupiska słabszych motywów konsensusu, być może w celu osiągnięcia podobnego obłożenia. Wiążące motywy CTCF wykorzystywały cztery „moduły”. Połowa powiązanych witryn CTCF korzystała z modułów 1 i 2, podczas gdy reszta korzystała z kombinacji tych czterech. Uważa się, że CTCF używa palców cynkowych do rozpoznawania różnych kombinacji tych modułów.
Rhee i Pugh przeanalizowali strukturę i organizację kompleksu przedinicjacyjnego (PIC) w genomach Saccharomyces . Korzystając z ChIP-exo, byli w stanie między innymi precyzyjnie zidentyfikować cechy podobne do TATA u promotorów, o których zgłoszono brak TATA.
Zobacz też
Linki zewnętrzne
- Interakcje DNA-białko w wysokiej rozdzielczości
- Rozwiązywanie wiązania czynnika transkrypcyjnego
- Immunoprecypitacja chromatyny w wysokiej rozdzielczości
- Ważne białka regulujące geny określone nową metodą
- CexoR: Pakiet R/Bioprzewodników do wykrywania interakcji białko-DNA o wysokiej rozdzielczości w replikacjach ChIP-exo
- Genomika pekoniczna