CAPP-Sekw
| Akronim | CAPP-Sekw |
|---|---|
| Używa | Kwantyfikacja niskiego poziomu ctDNA od pacjentów z rakiem. |
| Godne uwagi eksperymenty | CAPP-Seq zastosowano na niedrobnokomórkowym raku płuca (NSCLC) w celu zidentyfikowania nawracających zmian somatycznych z ctDNA. |
| Powiązane przedmioty | Bezkomórkowe DNA nowotworu |
P ersonalizowane profilowanie CA ncer przez głębokie sekwencjonowanie (CAPP-Seq) to oparta na sekwencjonowaniu metoda następnej generacji, stosowana do ilościowego oznaczania krążącego DNA w raku ( ctDNA ). Metoda została wprowadzona w 2014 roku przez laboratoria Asha Alizadeha i Maximiliana Diehna w Stanford jako narzędzie do pomiaru bezkomórkowego DNA nowotworu , które jest uwalniane z martwych komórek nowotworowych do krwi, a tym samym może odzwierciedlać cały genom guza. Metodę tę można uogólnić dla każdego typu raka, o którym wiadomo, że ma nawracające mutacje. CAPP-Seq może wykryć jedną cząsteczkę zmutowanego DNA na 10 000 cząsteczek zdrowego DNA. Oryginalna metoda została udoskonalona w 2016 r. w celu uzyskania ultraczułej detekcji poprzez integrację wielu strategii tłumienia błędów, określanych jako zintegrowane tłumienie błędów cyfrowych (iDES). Wykorzystania ctDNA w tej technice nie należy mylić z krążącymi komórkami nowotworowymi (CTC); to są dwa różne byty.
Pierwotnie opisany jako metoda wykrywania i monitorowania raka płuc, CAPP-Seq został pomyślnie dostosowany do szerokiego zakresu nowotworów przez wiele niezależnych grup. Należą do nich chłoniak rozlany z dużych komórek B (DLBCL), chłoniak grudkowy (FL), zespół limfoproliferacyjny po przeszczepie (PTLD), rak jelita grubego z przerzutami do jajnika , rak przełyku , rak trzustki , rak pęcherza moczowego , mięśniakomięsak gładkokomórkowy , różne mięsaki dorosłych i dzieci , pośród innych.
metoda
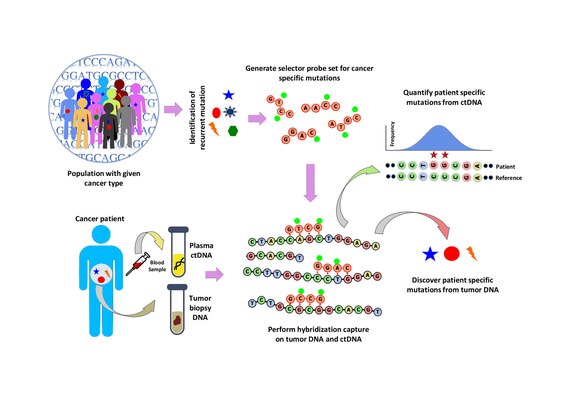
Analizę populacji przeprowadza się w celu identyfikacji powtarzających się mutacji w danym typie nowotworu. Odbywa się to poprzez analizę publicznych zbiorów danych, takich jak baza danych raka COSMIC i TCGA . Zaprojektowano „selektor”, który składa się z biotynylowanych sond oligonukleotydowych DNA ukierunkowanych na powtarzające się mutacje regionów wybranych dla konkretnego typu nowotworu. Selektor jest wybierany przy użyciu wielofazowego podejścia bioinformatycznego. Za pomocą selektora przeprowadza się wychwyt hybrydyzacyjny oparty na sondzie na guzie i normalnym DNA w celu wykrycia mutacji specyficznych dla pacjenta. Przechwycenie hybrydyzacji jest następnie stosowane również do ctDNA w celu ilościowego określenia mutacji, które zostały wcześniej odkryte.
Ekstrakcja ctDNA i przygotowanie biblioteki
osocza izoluje się ctDNA . DNA wejściowe może mieć zaledwie 4 ng.
Adaptacja tego protokołu do pracy z ctDNA miała cztery główne cele:
- 1) w celu optymalizacji wydajności ligacji adaptera
- 2) w celu zmniejszenia liczby cykli PCR potrzebnych po ligacji
- 3) aby zachować naturalnie występujący rozkład wielkości ctDNA (mediana 170 par zasad)
- 4) aby zminimalizować zmienność głębokości pokrycia sekwencji we wszystkich przechwyconych regionach
Osiągnięto to poprzez umożliwienie przeprowadzenia ligacji adaptera w temperaturze 16°C przez 16 godzin w celu zwiększenia wydajności ligacji adaptera i regeneracji. Najważniejsza adaptacja zachodzi podczas etapów enzymatycznych i oczyszczania; są one wykonywane z kulką, aby zminimalizować etapy przenoszenia rurki, co zwiększa regenerację.
Projekt selektora
W CAPP-Seq zaprojektowanie selektora jest kluczowym krokiem, który identyfikuje powtarzające się mutacje w określonym typie raka przy użyciu publicznie dostępnych danych sekwencjonowania nowej generacji. W celu włączenia do selektora CAPP-seq, powtarzające się mutacje, które są wzbogacane w populacji, są opisane za pomocą indeksu - Recurrence Index (RI). RI to liczba mutacji na kilozasady danego locus genomowego pacjenta niosącego określone mutacje. RI reprezentuje częstość nawrotów na poziomie pacjenta oszacowaną dla mutacji somatycznych i wszystkich mutacji. Znane i powtarzające się mutacje w populacji można uszeregować na podstawie RI, dlatego też RI jest używany do zaprojektowania selektora. Sześciofazowa strategia projektowania jest stosowana do selektora projektu.
- Faza 1: Identyfikacja częstych mutacji znanych mutacji kierowców przy użyciu publicznie dostępnych danych.
- Faza 2: Maksymalne pokrycie SNV wśród pacjentów zostało określone przez uszeregowanie ich eksonowego RI.
- Faza 3 i 4: Wybrano eksony o wyższym RI.
- Faza 5: Dodanie wcześniej przewidywanych mutacji kierowców.
- Faza 6: Dodanie powtarzających się rearanżacji fuzji genów, które są specyficzne dla konkretnego raka.
Ludzki rak jest heterogenny, a nawracające mutacje nowotworowe występują tylko u mniejszości pacjentów. Dlatego ostrożny i nieredundantny projekt selektora jest istotną częścią CAPP-Seq, a także rozmiar selektora jest związany z jego dalszymi kosztami.
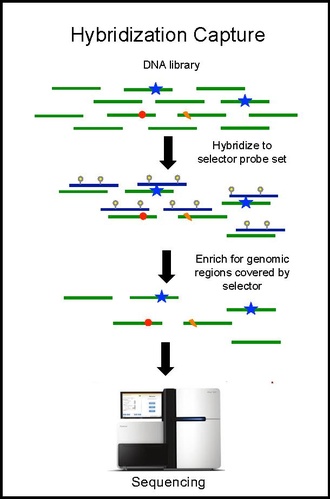
Przechwytywanie i sekwencjonowanie hybrydyzacji
Wychwytywanie hybrydyzacji za pomocą zestawu sond selektorowych przeprowadza się na DNA guza z biopsji i sekwencjonuje do głębokości pokrycia ~ 10 000x. Biotynylowane sondy selektorowe wiążą się selektywnie z regionami biblioteki DNA, które zostały wybrane jako miejsca występowania powtarzających się mutacji w danym typie nowotworu. W ten sposób pozostaje ci mniejsza biblioteka, która jest wzbogacona tylko o żądane regiony, które można następnie zsekwencjonować. Pozwala to na określenie specyficznych dla pacjenta mutacji. Następnie przeprowadza się wychwyt hybrydyzacji za pomocą tego samego selektora na ctDNA z krwi w celu ilościowego określenia wcześniej zidentyfikowanych mutacji u pacjenta. CAPP-Seq można zastosować do ctDNA z wielu próbek krwi w różnych punktach czasowych, aby śledzić ewolucję guza.
Potok obliczeniowy dla CAPP-seq
Analiza danych CAPP-Seq obejmuje szereg kroków, od wykrywania mutacji po walidację, a oprogramowanie typu open source może wykonać większość analizy. Po pierwszym etapie wywoływania wariantów mutacje linii zarodkowej i utraty heterozygotyczności (LOH) są usuwane z CAPP-seq w celu zmniejszenia błędów tła. Kilka testów istotności statystycznej można przeprowadzić na tle wszystkich rodzajów wywołań wariantowych. Na przykład istotność statystyczną SNV pochodzących z guza można oszacować przez losowe pobieranie próbek alleli tła przy użyciu metody Monte Carlo . W przypadku wywołań indel istotność statystyczna jest obliczana przy użyciu oddzielnej metody, która wykorzystywała analizę specyficzną dla nici za pomocą testu Z pokazanego w poprzedniej pracy. Wreszcie etapy walidacji obliczeniowej zmniejszają liczbę fałszywych alarmów. Jednak w tej dziedzinie istnieje duże zapotrzebowanie na solidne ramy obliczeniowe specyficzne dla analizy danych CAPP-seq.
Wrażliwość
Czułość tej technologii zależy od skutecznego projektu selektora i jest silnie obciążona wielkością kohorty i rodzajem badanego raka. Brak tła do znalezienia statystycznie istotnych powtarzających się wariantów ograniczył jego wydajność ze względu na szum stochastyczny i zmienność biologiczną. charakterystyki operacyjnej odbiornika (ROC) na kilku pacjentach z rakiem i pacjentach wyleczonych z raka (próbki pobrane w różnych stadiach nowotworu, punkt czasowy krążącego DNA, leczenie itp.) Wykazała, że CAPP-seq ma wyższą czułość i specyficzność w porównaniu z poprzednimi metodami w nie- drobnokomórkowy rak płuca.
Ograniczenia
Na granicę wykrywalności CAPP-Seq mają wpływ trzy główne obszary: wejściowa ilość cząsteczek ctDNA, zanieczyszczenie krzyżowe próbki, potencjalne obciążenie alleliczne w odczynniku wychwytującym oraz PCR lub błędy sekwencjonowania. ctDNA można wykryć przy dolnej granicy 0,025% ułamkowej obfitości we krwi. Stwierdzono, że zanieczyszczenie krzyżowe próbek miało bardzo mały wpływ, a raporty wykazały minimalne obciążenie alleliczne w kierunku wychwytywania alleli referencyjnych w PBL ( limfocytach krwi obwodowej ). Błędy PCR i sekwencjonowania są również minimalne. Technika staje się wątpliwa, gdy ctDNA jest obecny na niskim poziomie 0,01%. Ponadto, gdy jest mniej uwalniania ctDNA z powodu stabilności wzrostu guza przez terapię, wykrywanie jest zagrożone.
Nadal nie wiadomo, czy ctDNA jest uwalniane z równym czy nierównym tempem z guzów pierwotnych i chorób przerzutowych . Fakt ten należy wziąć pod uwagę podczas wykonywania CAPP-Seq, ponieważ może to powodować problemy w określaniu obciążenia nowotworem i ewolucji klonalnej, jeśli różne guzy lub klony obumierają i uwalniają swoje DNA w różnym tempie. Nie wiadomo również, w jaki sposób histologia guza wpływa na uwalnianie ctDNA.
Innym poważnym ograniczeniem używania tylko poziomów ctDNA do wykrywania obciążenia nowotworem jest to, że ctDNA może przewidywać jedynie guz resztkowy, nie może nic powiedzieć o lokalizacji guza. Oznacza to, że CAPP-Seq może być najlepiej stosowany jako uzupełnienie innych podejść do sekwencjonowania do obrazowania obciążenia chorobą w różnych momentach. W związku z tym techniczna wrażliwość, odtwarzalność, specyficzność i wymóg wiedzy specjalistycznej do analizy dużej ilości danych to tylko niektóre z problemów związanych z tą techniką.
Zalety
CAPP-Seq ma wiele zalet w porównaniu z innymi metodami, takimi jak cyfrowa reakcja łańcuchowa polimerazy (dPCR) i sekwencjonowanie amplikonów . CAPP-Seq może badać wiele loci w tym samym eksperymencie w porównaniu z sekwencjonowaniem dPCR i amplikonu, które wykorzystują wiele różnych eksperymentów i dlatego zużywają znacznie więcej próbek. Kolejną zaletą jest to, że CAPP-Seq może nie tylko wykrywać mutacje punktowe, ale może również wykrywać indele , zmiany strukturalne i zmiany liczby kopii , a także pomaga w monitorowaniu minimalnej choroby resztkowej.
Kolejną zaletą CAPP-Seq jest to, że ponieważ jest ukierunkowany tylko na określone obszary genomu, jest bardziej opłacalny niż sekwencjonowanie całego egzomu i sekwencjonowanie całego genomu , które są odpowiednio 171X i 44X droższe. Ponadto nie ma potrzeby dyskretnego usprawniania dla poszczególnych pacjentów.
Wykorzystanie krążącego DNA guza w przeciwieństwie do biopsji guza litego umożliwia analizę pełnego repertuaru komórek nowotworowych rozproszonych w obrębie guza i przerzutów odległych. Dlatego istnieje większa szansa na znalezienie wszystkich mutacji związanych z tym nowotworem. Posiadanie pełnego obrazu nowotworu i jego przyczyn pozwoli na lepsze planowanie leczenia i zarządzanie chorobą.
Aplikacje
Monitorowanie obciążenia nowotworem
W leczeniu raka przydatne są dokładne pomiary całkowitego obciążenia chorobą organizmu. Pomaga w określeniu znaczenia prognostycznego i odpowiedzi na leczenie. Zwykle wykonuje się to za pomocą tomografii komputerowej ( tomografia komputerowa ), pozytonowej tomografii emisyjnej ( skany PET ) lub rezonansu magnetycznego ( MRI ). Te procedury obrazowania medycznego są drogie i nie są pozbawione własnych problemów. Te techniki obrazowania nie są w stanie dokładnie rozdzielić małych guzów (o średnicy ≤1 cm). Na obrazowanie mogą również wpływać stany zapalne wywołane promieniowaniem i zmiany zwłóknieniowe, co utrudnia określenie, czy istnieje resztkowy guz, czy tylko efekty leczenia.
Stwierdzono, że poziomy ctDNA w osoczu istotnie korelują z objętością guza w porównaniu z obrazowaniem medycznym (CT, PET i MRI). Wykrywanie ctDNA może przewidzieć resztkowy guz lub rychły nawrót, w niektórych przypadkach nawet lepiej niż obrazowanie medyczne i obecne metody.
Wskaźnik prognostyczny
Jak dotąd w wielu badaniach stwierdzono, że wykrycie ctDNA jest predyktorem nawrotu. W badaniu dotyczącym późnego stadium NSCLC (niedrobnokomórkowego raka płuca) znaleźli dwa przypadki, w których ctDNA prawidłowo określiło wynik pacjenta, gdy obrazowanie medyczne było błędne. W jednym przypadku obrazowanie przewidywało nawrót na podstawie podejrzenia resztkowego guza, który okazał się jedynie zapaleniem wywołanym promieniowaniem, ale nie wykryto ctDNA i pacjent nie miał nawrotu. W innym przypadku obrazowanie nie wykazało guza, ale wykryto ctDNA i wkrótce potem doszło do nawrotu choroby. W innym badaniu dotyczącym DLBCL (rozlanego chłoniaka z dużych komórek B) stwierdzono również, że ctDNA może przewidywać nawrót choroby.
Genotypowanie guza bez biopsji
Biopsje są inwazyjne i wiążą się z ryzykiem dla pacjenta. Dlatego wielokrotne biopsje w celu monitorowania postępu choroby są rzadkie, a biopsje diagnostyczne są podstawą informacji genetycznej. Może to być problematyczne ze względu na niejednorodność guza i jego ewolucję. Po pierwsze, biopsja pobiera próbkę tylko z jednej części guza, a ponieważ guzy są heterogenne, nie obejmuje to pełnego krajobrazu genetycznego guza. Po drugie, po leczeniu nowotwory ewoluują i mogą pojawić się nowe mutacje niereprezentowane w próbce diagnostycznej.
Genotypowanie guza bez biopsji za pomocą CAPP-Seq i ctDNA rozwiązuje wiele z tych problemów. Proste badanie krwi jest nieinwazyjne, znacznie bezpieczniejsze i łatwiejsze w wielokrotnym poddawaniu pacjentów z rakiem w trakcie leczenia. Użycie ctDNA daje lepszą próbkę DNA guza w porównaniu z pojedynczym obszarem guza pobranym w biopsji, co pozwala na lepsze oszacowanie heterogeniczności guza. Pobranie wielu próbek ctDNA w różnych punktach czasowych po przebiegu leczenia umożliwia odkrycie ewolucji guza. Może to pomóc wykryć pojawienie się mutacji, które nadają oporność na terapię celowaną i umożliwić odpowiednie dostosowanie przebiegu leczenia. CAPP-Seq w szczególności pozwala na badanie przesiewowe wielu lokalizacji genomowych, które staną się ważne, ponieważ lista mutacji nowotworowych ważnych dla leczenia wciąż rośnie. W badaniu późnego stadium NSCLC przeprowadzili wersję CAPP-Seq, w której biopsja guza nie była najpierw sekwencjonowana, i byli w stanie poprawnie sklasyfikować 100% próbek osocza pacjentów z 0% odsetkiem wyników fałszywie dodatnich. Pokazuje to, że nawet bez wcześniejszej wiedzy o mutacjach nowotworowych można je dokładnie wykryć za pomocą samego ctDNA.