Bizmutyniden

Bizmutynidyny to klasa związków organobizmutowych , analogiczna do karbenów . Związki te mają ogólną postać R-Bi, z dwiema samotnymi parami elektronów na centralnym atomie bizmutu (I). Ze względu na niezwykle niską wartościowość i stopień utlenienia +1, większość bizmutynidenów jest reaktywna i niestabilna, chociaż w ostatnich dziesięcioleciach zarówno metale przejściowe , jak i wielokleszczowe chelatujące ligandy zasady Lewisa były stosowane do stabilizacji niskowartościowego bizmutu (I) centrum poprzez steryczne ochrona i darowizna π w roztworze lub w strukturach krystalicznych. Bizmutynideny stabilizowane zasadą Lewisa przyjmują singletowy stan podstawowy z obojętną samotną parą elektronów na orbicie 6s . Druga samotna para w orbicie 6p i pojedynczy pusty orbital 6p sprawiają, że bismuthinideny stabilizowane zasadą Lewisa są ambifilne.
Synteza
Bizmutyniden stabilizowany metalem przejściowym
Najwcześniejsze przykłady kompleksów bizmutynidenu wykorzystywały chemię metali przejściowych do stabilizacji centrum Bi (I). Metody te ogólnie wykorzystywały zdolność prostych halogenków bizmutu (I) lub metylobizmutu do ligacji z kompleksami karbonylu wolframu , manganu i chromu . Czasami stwierdzono, że kompleksy te oligomeryzują , tworząc pojedyncze lub podwójne wiązania Bi-Bi, tworząc ugrupowania bizmutanowe lub bizmutenowe . Jeden z pierwszych przykładów monomerycznego bizmutynidenu został odkryty przez Balasza i wsp., którzy użyli R = 2-(dimetyloaminometylo)fenylu do chelatowania centrum Bi(I) poprzez połączenie silnych oddziaływań C-Bi i słabych N-Bi. Chociaż cząsteczka szybko utworzyła cykliczny oligomer, w reakcji z dwoma równoważnikami pentakarbonylu wolframu wyodrębniono monomeryczny krystaliczny RBi[W(CO) 5 ] 2 .

N , C , N -skoordynowany bizmutyniden
Monomeryczne bizmutynideny nie były stabilizowane bez użycia kompleksów metali przejściowych aż do 2010 roku, kiedy grupa badawcza Libor Dostál doniosła o wyizolowaniu centrum bizmutu(I) stabilizowanego jedynie przez N , C , N - pincer ligand L = 2,6 - bis [ N- (2',6'-dimetylofenylo)ketimino]fenyl. Kompleks ten został najpierw zsyntetyzowany przez reakcję cząsteczki prekursora LBi III Cl 2 z dwoma równoważnikami czynnika redukującego K[B( iBu ) 3H ] w celu uzyskania izolowanych kryształów stabilnego [C 6H 3-2,6- (C( Me)=N-2′, 6′ - Me2C6H3 ) 2 ] Bi . Nieco prostszy ligand koordynujący N , C , N został wkrótce użyty do stworzenia bizmutynidenu ArBi (Ar = C 6 H 3 -2,6-(CH=N t Bu) 2 ), który stał się szeroko stosowany w późniejszych badaniach nad bizmutynidenem i jest czasami określany jako „bismuthinidene Dostála”. W rzeczywistości zsyntetyzowano wiele analogów tego związku, często zastępując grupy tert -butylowe iminy innymi dużymi grupami organicznymi lub zastępując dwa ramiona iminowe ramionami dipodstawionej aminy.

N , C -skoordynowany bizmutyniden
Grupa Dostála zsyntetyzowała później monomeryczny bizmutyniden koordynowany tylko przez dwukleszczowy ligand koordynujący N , C. Gdy dichlorek bizmutu [C 6 H 2 -2-(CH=N-2',6'- i Pr 2 C 6 H 3 )-4,6-( t Bu) 2 ]BiCl 2 zostanie zredukowany o dwa równoważniki K[B( i Bu) 3 H], izolowane ciemnofioletowe kryształy [C 6 H 2 -2-(CH=N-2',6'- i Pr 2 C 6 H 3 )-4,6-( t Pojawia się Bu) 2 ]Bi. W przeciwieństwie do wcześniejszego stabilizowanego metalem przejściowym [2-(Me 2 NCH2)C 6 H 4 ]Bi[W(CO) 5 ] 2 , grupa tert -butylowa orto do atomu bizmutu w tym N , C -skoordynowanym bizmutynidenie sterycznie blokują częściowo pusty orbital typu p na atomie bizmutu, stabilizując go kinetycznie bez użycia metali przejściowych. Ponadto obliczone niezależne od jądra indeksy przesunięcia chemicznego (NICS) oraz analiza anizotropii gęstości indukowanej prądem (ACID) pokazują, że pierścień BiC3 N cząsteczki został ustabilizowany w pewnym stopniu dzięki charakterowi aromatycznemu i może być sklasyfikowany jako benzazabismol do delokalizacji sześciu elektronów π, pomimo nominalnie celowniczego wiązania Bi-N. W przeciwieństwie do bizmutynidenów skoordynowanych N, C, N, ten skoordynowany gatunek N, C wymaga, aby boczny atom azotu znajdował się w grupie iminowej, ponieważ zastąpienie ramienia iminowego podstawionego Dipp ramieniem aminowym podstawionym dietylem spowodowało szybką dimeryzację do gatunek dibizmutenu.

Bizmutyniden stabilizowany karbenem
W 2019 roku Wang i in. wyizolowali nowy bizmutyniden stabilizowany karbenem z egzocyklicznym centrum bizmutu (I). Dichlorek fenylobizmutu, stabilizowany cyklicznym alkiloaminokarbenem podstawionym dietylo/diizopropylofenylem ( Et2 CAAC), reaguje z jednym równoważnikiem kompleksu berylu (0) Be ( Et2 CAAC) 2 w toluenie, dając stabilne, dające się wyodrębnić czerwone kryształy karbeno- stabilizowany bizmutyniden ( Et2CAAC )Bi-Ph. Pomimo odsłoniętego, egzocyklicznego centrum bizmutu(I), związek ten może istnieć bez dimeryzacji do dwóch tygodni w stanie stałym. z teorii funkcjonału gęstości (DFT) wykazały, że jest to wynikiem częściowego charakteru wiązania podwójnego między węglem karbenu a centrum bizmutu(I), w którym samotna para elektronów typu p na atomie bizmutu oddziałuje z częściowo wypełnionym p orbitale na węglu karbenowym. Jednak ładunek atomu bizmutu określony za pomocą analizy naturalnej populacji (NPA) był znacznie niższy niż w wiązaniach bizmut (III)-węgiel, co potwierdza klasyfikację związku jako bizmutynidenu.

Właściwości strukturalne
Podobnie jak w przypadku innych cięższych analogów karbenu, właściwości strukturalne i elektroniczne bizmutynidenów są w dużej mierze napędzane efektem pary obojętnej , w którym duża przerwa energetyczna między orbitalami 6s i 6p atomu bizmutu nie sprzyja tworzeniu orbitali hybrydowych sp . W przeciwieństwie do ich lżejszych kongenerów fosfinidenów , których mniejsza przerwa energetyczna 3s-3p fosforu sprzyja trypletowemu stanowi podstawowemu, bizmutynideny generalnie mają singletowy stan podstawowy ze względu na większą przerwę energetyczną bizmutu 6s-6p.
Strukturalne i elektroniczne właściwości bizmutynidenów w ogólności są wyraźnie zilustrowane przez N, C, N-stabilizowany bizmutyniden Dostála, który jest najczęściej stosowanym bizmutynidenem w dotychczasowej literaturze. Optymalizacja tert -butyloiminowej tego związku na poziomie teorii M06/cc-pVTZ ujawnia, że podobnie jak w innych nietrygonalnych związkach pniktogenu , centralny atom bizmutu jest współpłaszczyznowy z ligandem koordynującym N,C,N, przyjmując Tryb koordynacji C 2v w kształcie litery T. Indeks wiązania Wiberga (WBI) między bizmutem a atomami węgla wynosi 1,09, podczas gdy odległość wiązania Bi-C wynosi 2,2156 Å, nieco mniej niż suma promieni kowalencyjnych pojedynczych wiązań tych atomów (Σ cov (Bi, C) = 2,26 Å). Z drugiej strony WBI wiązań Bi-N wynosi tylko 0,34, a odległość między wiązaniami Bi-N wynosi 2,500 Å, czyli znacznie więcej niż suma promieni kowalencyjnych tych atomów (Σ cov (Bi,N) = 2,22 Å ). Jest to zgodne z obliczeniami opartymi na kwantowej teorii atomów w cząsteczkach (QTAIM), które pokazują, że gęstość elektronów w punkcie krytycznym wiązania między Bi i N wynosi tylko 0,049, znacznie mniej niż gęstość elektronów wynosząca 0,114 w wiązaniu Bi-C punkt krytyczny. Obliczenia orbitalu wiązań naturalnych (NBO) pokazują, że te słabsze wiązania celownicze wynikają ze słabego oddawania σ samotnych par atomów azotu na pusty orbital 6p na centralnym atomie bizmutu. Te dwa oddziaływania n N → p* Bi stabilizują bizmutyniden aż o 382 kJ/mol. Dodatkowo, ilość donacji sigma z bocznych atomów azotu można zwiększyć lub zmniejszyć przez zastąpienie grup tert -butylowych na bocznych atomach azotu grupami arylowymi zawierającymi odpowiednio grupy elektronodonorowe lub grupy elektronoakceptorowe. Jedna samotna para znajduje się na orbicie 6s atomu bizmutu i generalnie pozostaje obojętna, podczas gdy druga znajduje się na orbicie 6p zorientowanej prostopadle do płaszczyzny pierścienia centralnego, który obejmuje również najwyższy zajęty orbital molekularny (HOMO).
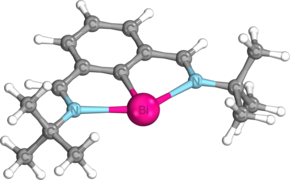
Zoptymalizowana geometria bizmutynidenu Dostála

samotna para typu p na Bi
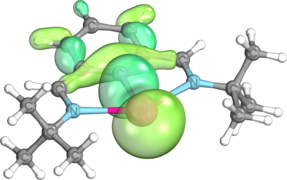
σ-wiązanie między Bi i C

σ-donacja N samotnych par na pusty orbital typu p na Bi
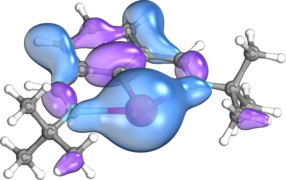
samotna para typu s na Bi





Reaktywność
Teoretycznie bizmutynideny są zarówno kwaśne, jak i zasadowe Lewisa ze względu na odpowiednio puste i wypełnione orbitale typu p. W praktyce bizmutynideny chelatowane zarówno N, C, N, jak i N, C tracą wiele ze swojego kwasowego charakteru Lewisa z powodu interakcji donor-akceptor n N → p * Bi . Jednak zasadowość Lewisa bizmutynidenów, zwłaszcza bizmutynidenu koordynowanego przez N, C, N Dostála, pozwala im na przewidywalne cykle między stabilnymi stanami utlenienia Bi (I) i Bi (III) w zależności od warunków reakcji, pozwalając im działać jako katalizatory dla szereg różnych reakcji, w tym uwodornienia z przeniesieniem , deoksygenacje , hydrodefluoracje i redukcja diwodoru. Ponadto bizmutynideny reagują samoistnie z niektórymi halogenkami alkilowymi, dichalkogenkami i alkinami, tworząc związki Bi (III).

Reaktywność katalityczna
Stosunkowo duży promień kowalencyjny bizmutu powoduje słabsze wiązania między bizmutem a innymi pierwiastkami. Jest to szczególnie prawdziwe w przypadku bizmutynidenów koordynowanych przez N, C, N po oksydacyjnym dodaniu dodatkowych ligandów w celu utworzenia reaktywnych związków pośrednich z trójwartościowym bizmutem (III), biorąc pod uwagę darowiznę n N → p * Bi obecną w tych gatunkach. Ta względna słabość wiązań bizmutu (III) z ligandem ułatwia wymianę ligandów i ostatecznie redukcyjną eliminację w celu uwolnienia produktu, zreformowania bizmutynidenu i stworzenia kontrolowanego cyklu redoks Bi (I) / Bi (III), który nadaje bizmutynidenom ich własne unikalne właściwości katalityczne reaktywność.
Uwodornianie transferowe azoarenów
W 2019 roku Wang i in. , który wykorzystał aktywność katalityczną bizmutynidenu Dostála do katalizowania reakcji uwodornienia przeniesienia między amoniakiem-boranem i azoarenami , tworząc odpowiednie arylohydrazyny z dobrą tolerancją grup funkcyjnych. Cykl katalityczny reakcji przebiega poprzez utleniające dodanie dwóch atomów wodoru z amoniaku-borowodoru do centrum bizmutu (I), tworząc wysoce niestabilny bizmutynowy związek pośredni. Późniejsza eliminacja redukcyjna przenosi dwa atomy wodoru przez wiązanie pi cząsteczki azoarenu, przywracając bizmutyniden i tworząc arylohydrazynę. Podobna reakcja uwodornienia z przeniesieniem katalizowana bizmutynidenem redukuje nitroareny do odpowiednich arylohydroksyloamin.
Odtlenienie podtlenku azotu

Para redoks Bi (I) / Bi (III) została również zastosowana do katalizowania odtlenienia podtlenku azotu . Kiedy bizmutyniden Dostála jest wystawiony na działanie gazowego N2O , mieszanina reakcyjna zmienia kolor z zielonego na żółty i wydziela się gazowy azot. Zmiana koloru jest spowodowana utworzeniem dimeru tlenku arylobizmutu z dwoma μ -okso tworzącymi centrum Bi 2 O 2 , zgodnie ze skłonnością tlenków bizmutu(III) do spontanicznej dimeryzacji lub polimeryzacji. Jednak zmodyfikowana wersja bizmutynidenu Dostála z ramionami ketiminowymi i m -terfenylowymi na atomach azotu ketiminy nie sprzyja dimeryzacji, zamiast tego tworzy rzadki monomeryczny wodorotlenek organobizmu(III) w reakcji z N2O . W obu przypadkach redukcja produktu za pomocą pinakoboran (HBpin) przywraca centra bizmutu(III) do stanu bizmutu(I) i daje mieszaninę HOBpin i (pinB) 2O , kończąc cykl katalityczny.
Hydrodefluorowanie polifluoroarenów
Właściwości elektroniczne ligandu N, C, N-pincer można dostroić za pomocą grup odciągających elektrony, aby promować reaktywność bizmutynidenów w kierunku wiązań arylowych CF. Jednym z przykładów jest Phebox-Bi (I), N, C, N-koordynowany bizmutyniden stabilizowany 2,6-bis (oksazolinylo) fenylo (Phebox) ligandem pincerowym. W przeciwieństwie do bizmutynidenu Dostála, który wykazał reaktywność tylko w stosunku do pentafluoropirydyny, Phebox-Bi (I) wykazał skłonność do dodawania do wiązań CF w różnych perfluorowanych arenach, w tym pentafluoropirydynie, podstawionych pentafluorobenzenach, wysoce fluorowanych związkach fosfinowych, oktafluoronaftalenie i dekafluorobifenylu. Po utleniającym dodaniu do wiązania arylowego CF, powstały związek pośredni Phebox-Bi(III)(fluoroarylo)fluorek może ulec metatezie ligandu z dietylosilanem, zastępując wiązanie Bi(III)-F wiązaniem Bi(III)-H. Niestabilny wodorek Bi(III) przechodzi następnie redukcyjną eliminację arylu CH, regenerując Phebox-Bi(I) i hydroodfluorowany produkt. Katalizator zwykle celuje w wiązania CF para do dowolnych podstawników lub heteroatomów odciągających elektrony na fluorowanym podłożu. Szybkości reakcji znacznie spadają, gdy fluorowane podłoże zawiera grupę elektronodonorową.
Redukcja diwodoru z kwasu octowego
Badania elektrochemiczne wskazują, że bizmutyniden Dostála może służyć jako elektrokatalizator do tworzenia gazowego wodoru. Obliczenia woltamperometrii cyklicznej i DFT wskazują, że w warunkach redukujących kwas octowy wiąże się z centralnym atomem bizmutu, przenosząc atom wodoru do bizmutu i wytwarzając pośredni octanowodorek bizmutu (III) koordynowany przez N, C, N. Przegrupowanie ligandu octanowego z pozycji równikowej do osiowej pozwala drugiemu równoważnikowi kwasu octowego związać się z centrum bizmutu, eliminując przy tym H2 . Redukcja dwuelektronowa uwalnia ligandy octanowe, regenerując katalizator bizmutynidenowy.
Reaktywność wewnętrzna
Wewnętrzne reakcje transformacji bizmutynidenu na ogół obejmują podwójne utlenianie ze stanu redoks bizmutu (I) do stanu redoks bizmutu (III), generując niesymetrycznie podstawione trójwartościowe związki bizmutu (III), które byłyby trudne do syntezy za pomocą odczynników metaloorganicznych.
Addycja utleniająca w kierunku halogenków alkilowych i difenylodichalkogenków
Niska wartościowość bizmutynidenów powoduje, że są one reaktywne w stosunku do wiązań grupowych z polaryzacją węgla. Utleniające reakcje addycji między bizmutynidenem Dostála a pierwszorzędowymi wiązaniami C(sp3 ) -X są szczególnie korzystne dla X = I lub OTf, przekształcając centrum bizmutu(I) w halogenek bizmutu(III) alkilu lub triflat alkilu . Dotyczy to nawet dłuższych fluorowanych halogenków alkilowych o długości do sześciu atomów węgla. Zawada przestrzenna zapobiega aktywacji jodku tert -butylu przez bizmutyniden Dostála, chociaż metastabilny analog z aminowymi ramionami szczypiec zamiast iminowymi ramionami szczypiec (Ar'Bi, gdzie Ar' = 2,6-C 6 H 3 (CH 2 NMe 2 ) 2 ) uczestniczy w addycji utleniającej nawet z masywnymi trzeciorzędowymi wiązaniami CX, prawdopodobnie dlatego, że zwiększona ruchliwość obrotowa ramion aminowych pozwala im obracać się z dala od nadchodzącej masywnej grupy alkilowej w stanie przejściowym.
Ten metastabilny Ar'Bi, jak również bizmutyniden stabilizowany N,C, są również reaktywne wobec difenylodichalkogenków. Podczas gdy pierwsza daje stabilne kryształy Ar'Bi(III)(EPh) 2 (E = S, Se, Te) w reakcji z PhEEPh, druga daje [2-C 6 H 4 (CH=NC 6 H 3 ( i -Pr) 2-2,6 )] 2Bi (III)(EPh) z dwoma ligandami N,C i tylko jednym chalkogenolanem fenylu. N,C,N- i podwójnie N,C-skoordynowane fenylotellulany bizmutu(III) są szczególnie nietrwałe i rozkładają się, tworząc mieszaninę produktów. Utleniające addycje N, C, N-skoordynowanego bizmutynidenu do dwusiarczków diarylu są tolerancyjne wobec różnych arylowych grup funkcyjnych, w tym grup pirydylowych, tiazolilowych, tienylowych i aminofenylowych.

Reakcja Hetero Dielsa-Aldera z alkinami
W 2019 roku Kořenková i in. odkrył, że bizmutyniden Dostála zachowuje się jak zamaskowany heterocykliczny dien w obecności alkinu dimetyloacetylenodikarboksylanu (DMAD) z niedoborem elektronów, przeprowadzając reakcję cykloaddycji hetero Dielsa-Aldera [4+2], dając CO 2 Me-dipodstawiony 1-bisma-1 ,4-dihydro-iminonaftalen, skutecznie przekształcając jedno z wiszących ramion iminowych bizmutynidenu w bismacykloheksadien z mostkiem azotowym, przy czym atom bizmutu (III) służy jako przyczółek, a drugie ramię iminy w dużej mierze traci koordynację z bizmutem (III) Centrum. Podobna reakcja cykloaddycji między bizmutynidenem Dostála i propiolanem metylu daje iminonaftalen tylko jako związek pośredni, ponieważ przyczółkowy atom bizmutu jest szybko atakowany przez deprotonowany drugi równoważnik propiolanu metylu, zrywając wiązanie Bi-N i dając ugrupowanie bismacykloheksadienu w kształcie łodzi . Chociaż technicznie nie jest mostkowana, osiowa grupa aminowa w pierścieniu bismacykloheksadienu pozostaje celowo skoordynowana z heteroatomem bizmutu.
Chemia metali przejściowych

Chociaż bizmutynideny koordynowane przez N, C, N są stabilne bez koordynacji z metalem przejściowym, są one reaktywne w stosunku do pewnych kompleksów metali przejściowych z niedoborem elektronów i działają jako ligandy donorowe typu L. Po dodaniu bizmutynidenu Dostála (ArBi; Ar = C 6 H 3 -2,6-(CH=N t Bu) 2 ) do roztworów oktakarbonylu dikobaltu lub dekakarbonylu dimanganu w toluenie, wyodrębniają się kryształy jonowe [(ArBi) 2 Co ( CO) 3 ] + [Co(CO) 4 ] - lub [(ArBi) 2 Mn(CO) 4 ] + [Mn(CO) 5 ] - zaczynają się tworzyć. Kompleksy te wykazują znaczące oddziaływanie kowalencyjne między atomami bizmutu (I) a kobaltu lub manganu, chociaż te wiązania bizmut-metal mają charakter celownika. Ponieważ wiązanie bizmut-metal składa się prawie wyłącznie z σ-oddawania elektronów w samotnej parze typu p na bizmucie do orbitalu dz 2 centrum metalu przejściowego, jednostki ArBi wiążą się z centrum metalu w bok sposób z kątami wiązania C-Bi-Co lub C-Bi-Mn bliskimi 90°, tak że płaszczyzny ligandów N,C,N i trygonalnej płaskiej Co(CO) 3 lub kwadratowej płaskiej Mn ( CO ) Wszystkie 4 ugrupowania są prawie równoległe do siebie. W tym trybie wiązania pierścienie aromatyczne ligandów N,C,N przyjmują syn stabilizowaną słabymi oddziaływaniami CH···CH i CH···O.
Bizmutyniden Dostála wiąże się również z centrami złota (I) stabilizowanymi przez N-heterocykliczny ligand karbenowy IPr (1,3-bis (2,6-diizopropylofenylo) imidazolin-2-yliden). Wymiana liganda z [Au(IPr)(ACN)] + [BF 4 ] - daje kompleks [Au(IPr)(ArBi)] + [BF 4 ] - w postaci białego proszku. Chociaż ten kompleks jest stabilizowany przez celowe oddanie elektronów z Bi(I) na pusty orbital 6s Au(I), to oddziaływanie Bi(I) → Au(I) jest jednak silniejsze niż jakiekolwiek wcześniej odkryte Bi(III) → Oddziaływania Au(I), w których donorem jest atom bizmutu. Ponadto reprezentuje pierwszy stabilny, izolowany kompleks zawierający oddziaływanie Bi(I) → Au(I), które uważa się za możliwe dzięki szkieletowi ligandu N, C, N-pincer.
Podobne reaktywności zaobserwowano również dla metastabilnej wersji bismutynidenu Dostála zawierającego ramiona szczypiec aminowych zamiast ramion szczypiec iminowych (Ar'Bi; Ar' = C 6H 3-2,6- ( CH 2NMe 2 ) 2 ) . Dodatek tego metastabilnego bizmutynidenu do THF M(CO) 5 (gdzie M = Cr, Mo , W) daje wyodrębnialne kryształy [Ar'BiM(CO) 5 ]. Tak jak poprzednio, jednostka Ar'-Bi wiąże się z ugrupowaniem M(CO) 5 w dz2 sposób side-on, z darowizną σ z Bi(I) do orbitalu metalu . Reakcja między Ar'-Bi i diironononakarbonylem również daje głównie [Ar'BiFe(CO) 5 ] wraz z niewielką ilością [Ar'Bi(Fe(CO) 4 ) 2 ] jako produkt drugorzędny. W rzeczywistości reaktywność bizmutynidenu Dostála ArBi i jego metastabilnego analogu Ar'-Bi w stosunku do metali przejściowych jest tak podobna, że w reakcji z Co 2 (CO) 8 tworzą one analogiczne kompleksy [(ArBi) 2 Co(CO) 3 ] + [Co(CO) 4 ] - i [(Ar'Bi) 2 Co(CO) 3 ] + [Co(CO) 4 ] - , odpowiednio, z podobnymi trybami wiązania, długościami wiązań i kątami wiązań.