PMS2
| PMS2 | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
 | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| Identyfikatory | |||||||||||||||||||||||||||||||||||||||||||||||
| , HNPCC4, PMS2CL, PMSL2, MLH4, PMS1 homolog 2, składnik systemu naprawy niedopasowań, MMRCS4, PMS-2 | |||||||||||||||||||||||||||||||||||||||||||||||
| Identyfikatory zewnętrzne | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||
| Wikidane | |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||||||||||||



Endonukleaza naprawy niedopasowanych zasad PMS2 jest enzymem , który u ludzi jest kodowany przez gen PMS2 .
Funkcjonować
Ten gen jest jednym z członków rodziny genów PMS2, które znajdują się w klastrach na chromosomie 7. Ludzkie geny związane z PMS2 znajdują się w prążkach 7p12, 7p13, 7q11 i 7q22. Egzony od 1 do 5 tych homologów mają wysoki stopień identyczności z ludzkim PMS2. Produkt tego genu bierze udział w naprawie niedopasowania DNA . Białko tworzy heterodimer z MLH1 i ten kompleks oddziałuje z MSH2 związanym z niedopasowanymi zasadami. Defekty tego genu są związane z dziedzicznym rakiem jelita grubego niezwiązanym z polipowatością , z zespołem Turcota i są przyczyną nadnamiotowego prymitywne guzy neuroektodermalne . Zaobserwowano alternatywnie składane warianty transkryptu.
Naprawa niedopasowania i aktywność endonukleazy
PMS2 bierze udział w naprawie niedopasowania i wiadomo, że ma ukrytą aktywność endonukleazy , która zależy od integralności motywu meta-wiązania w homologach MutL. Jako endonukleaza, PMS2 wprowadza nacięcia do nieciągłej nici DNA.
Interakcje
Wykazano, że PMS2 oddziałuje z MLH1 , tworząc heterodimer MutLα. Istnieje konkurencja między MLH3, PMS1 i PMS2 o domenę oddziałującą na MLH1, która znajduje się w resztach 492-742.
Oddziałujące domeny w PMS2 mają heptadowe powtórzenia, które są charakterystyczne dla białek zamka leucynowego. MLH1 oddziałuje z PMS2 przy resztach 506-756.
Heterodimery MutS, MutSα i MutSβ, łączą się z MutLα po wiązaniu z niedopasowaniem. Uważa się, że MutLα łączy etap rozpoznawania niedopasowań z innymi procesami, w tym: usuwaniem niedopasowań z nowej nici DNA, resyntezą zdegradowanego DNA i naprawą nacięcia w DNA. Wykazano, że MutLα ma słabą aktywność ATPazy, a także wykazuje aktywność endonukleazy, która wprowadza nacięcia do nieciągłej nici DNA. Ułatwia to degradację 5' do 3' niedopasowanej nici DNA przez EXO1. Miejsce aktywne MutLα znajduje się na podjednostce PMS2. PMS1 i PMS2 konkurują o interakcję z MLH1. Białka w interakcjom PMS2 zostały zidentyfikowane przez tandemowe oczyszczanie powinowactwa.
Ludzki PMS2 ulega ekspresji na bardzo niskim poziomie i nie uważa się, aby był silnie regulowany cyklem komórkowym.
Interakcje z udziałem p53 i p73
Wykazano również, że PMS2 oddziałuje z p53 i p73 . Pod nieobecność p53, komórki z niedoborem PMS2 i PMS2 sprawne są nadal zdolne do zatrzymania cyklu komórkowego w punkcie kontrolnym G2/M, gdy są traktowane cisplatyną . Komórki z niedoborem p53 i PMS2 wykazują zwiększoną wrażliwość na czynniki przeciwnowotworowe. PMS2 jest ochronnym mediatorem przeżycia komórek w komórkach z niedoborem p53 i moduluje ochronne szlaki odpowiedzi na uszkodzenia DNA niezależnie od p53. PMS2 i MLH1 mogą chronić komórki przed śmiercią komórki, przeciwdziałając apoptozie za pośrednictwem p73 w sposób zależny od naprawy niedopasowania.
PMS2 może wchodzić w interakcje z p73 w celu zwiększenia apoptozy indukowanej cisplatyną poprzez stabilizację p73. Cisplatyna stymuluje interakcję między PMS2 i p73, która jest zależna od c-Abl. Kompleks MutLα może działać jako adapter do przenoszenia p73 do miejsca uszkodzonego DNA, a także działać jako aktywator p73, ze względu na obecność PMS2. Możliwe jest również, że nadekspresja PMS2 stymuluje apoptozę pod nieobecność MLH1 iw obecności p73 i cisplatyny z powodu stabilizującego działania PMS2 na p73. Po uszkodzeniu DNA p53 indukuje zatrzymanie cyklu komórkowego przez p21 /WAF i inicjuje naprawę poprzez ekspresję MLH1 i PMS2. Kompleks MSH1/PMS2 działa jako czujnik rozmiaru uszkodzenia DNA i inicjuje apoptozę poprzez stabilizację p73, jeśli uszkodzenie jest nie do naprawienia. Utrata PMS2 nie zawsze prowadzi do niestabilności MLH1, ponieważ może on również tworzyć kompleksy z MLH3 i PMS1.
Znaczenie kliniczne
Mutacje
PMS2 to gen kodujący białka naprawy DNA zaangażowane w naprawę niedopasowania . Gen PMS2 znajduje się na chromosomie 7p22 i składa się z 15 eksonów. Ekson 11 genu PMS2 ma powtórzenie kodujące ośmiu adenozyn.
Kompleksowe profilowanie genomowe 100 000 próbek ludzkiego raka ujawniło, że mutacje w regionie promotora PMS2 są istotnie związane z dużym obciążeniem mutacyjnym guza (TMB), szczególnie w przypadku czerniaka . Wykazano, że TMB jest wiarygodnym predyktorem tego, czy pacjent może odpowiedzieć na immunoterapię przeciwnowotworową , gdzie wysoki TMB wiąże się z korzystniejszymi wynikami leczenia.
Heterozygotyczne mutacje linii zarodkowej w genach naprawy niedopasowania DNA, takich jak PMS2, prowadzą do autosomalnego dominującego zespołu Lyncha. Tylko 2% rodzin z zespołem Lyncha ma mutacje w genie PMS2. Wiek pacjentów, u których po raz pierwszy wystąpił zespół Lyncha związany z PMS2, jest bardzo zróżnicowany i wynosi od 23 do 77 lat.
W rzadkich przypadkach defekt homozygotyczny może powodować ten zespół. W takich przypadkach dziecko dziedziczy mutację genu od obojga rodziców, a stan ten nazywany jest zespołem Turcota lub konstytucyjnym niedoborem MMR (CMMR-D). Do 2011 roku zgłoszono 36 pacjentów z guzami mózgu spowodowanymi biallelicznymi mutacjami germinalnymi PMS2. Dziedziczenie zespołu Turcota może być dominujące lub recesywne. Recesywne dziedziczenie zespołu Turcota jest spowodowane złożonymi heterozygotycznymi mutacjami w PMS2. 31 z 57 rodzin zgłoszonych z CMMR-D ma mutacje germinalne PMS2. 19 z 60 homozygotycznych lub złożonych heterozygotycznych nosicieli mutacji PMS2 miało raka przewodu pokarmowego lub gruczolaki jako pierwszą manifestację CMMR-D. Obecność pseudogenów może powodować zamieszanie przy identyfikacji mutacji w PMS2, prowadząc do fałszywie pozytywnych wniosków na temat obecności zmutowanego PMS2.
Niedobór i nadmierna ekspresja
Nadekspresja PMS2 skutkuje hipermutacją i tolerancją na uszkodzenia DNA. Niedobór PMS2 również przyczynia się do niestabilności genetycznej, umożliwiając propagację mutacji z powodu zmniejszonej funkcji MMR. Wykazano, że myszy PMS2-/- rozwinęły chłoniaki i mięsaki. Wykazano również, że samce myszy z PMS2-/- są bezpłodne, co wskazuje, że PMS2 może odgrywać rolę w spermatogenezie.
Rola w normalnej okrężnicy
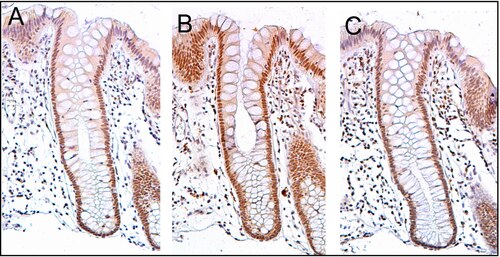
PMS2 jest zwykle wyrażany na wysokim poziomie w jądrach komórkowych enterocytów (komórkach absorpcyjnych) w kryptach okrężnicy wyściełających wewnętrzną powierzchnię okrężnicy ( patrz ilustracja, panel A). Naprawa DNA, obejmująca wysoką ekspresję białek PMS2, ERCC1 i ERCC4 (XPF), wydaje się być bardzo aktywna w kryptach okrężnicy w prawidłowym, nienowotworowym nabłonku okrężnicy. W przypadku PMS2 poziom ekspresji w prawidłowym nabłonku okrężnicy jest wysoki w 77% do 100% krypt.
Komórki są wytwarzane u podstawy krypty i migrują w górę wzdłuż osi krypty, zanim zostaną wydalone do światła okrężnicy kilka dni później. U podstawy krypt znajduje się od 5 do 6 komórek macierzystych . Jeśli komórki macierzyste u podstawy krypty wyrażają PMS2, generalnie wszystkie kilka tysięcy komórek krypty będzie również wyrażać PMS2. Wskazuje na to brązowy kolor widoczny w barwieniu immunologicznym PMS2 w większości enterocytów w krypcie w panelu A obrazu w tej sekcji. Podobna ekspresja ERCC4 (XPF) i ERCC1 występuje w tysiącach enterocytów w każdej krypcie okrężnicy normalnego nabłonka okrężnicy.
Sekcja tkanki na pokazanym tutaj obrazie była również barwiona kontrastowo hematoksyliną, aby zabarwić DNA w jądrach na niebiesko-szary kolor. Jądra komórek w blaszce właściwej (komórki, które znajdują się poniżej i otaczają krypty nabłonkowe) w dużej mierze wykazują niebieskoszary kolor hematoksyliny i wykazują niewielką ekspresję PMS2, ERCC1 lub ERCC4 (XPF).
Rak jelita grubego
Około 88% komórek pochodzenia nabłonkowego w raku okrężnicy i około 50% krypt okrężnicy w nabłonku w odległości 10 cm przylegających do raków (w ubytkach terenowych, z których prawdopodobnie powstały nowotwory) ma obniżoną lub nieobecną ekspresję PMS2.
Niedobory PMS2 w nabłonku okrężnicy wydają się wynikać głównie z represji epigenetycznej . W nowotworach sklasyfikowanych jako niedostateczne i pozbawione naprawy niedopasowania, w większości ekspresja PMS2 jest niewystarczająca z powodu braku partnera parowania MLH1 . Parowanie PMS2 z MLH1 stabilizuje się. Utrata MLH1 w sporadycznych nowotworach była spowodowana epigenetycznym spowodowanym metylacją promotora w 65 z 66 przypadków. W 16 nowotworach stwierdzono niedobór Pms2, mimo obecności ekspresji białka MLH1. Spośród tych 16 przypadków nie ustalono przyczyny dla 10, ale stwierdzono, że 6 miało heterozygotyczną mutację linii zarodkowej w Pms2, po której nastąpiła prawdopodobna utrata heterozygotyczności w guzie. Zatem tylko 6 ze 119 guzów pozbawionych ekspresji Pms2 (5%) było spowodowanych mutacją PMS2.
Koordynacja z ERCC1 i ERCC4 (XPF)

Kiedy PMS2 jest zredukowane w kryptach okrężnicy w ubytku pola, jest to najczęściej związane ze zmniejszoną ekspresją enzymów naprawy DNA ERCC1 i ERCC4 (XPF) (patrz obrazy w tej sekcji). Niedobór ERCC1 i/lub ERCC4 (XPF) spowodowałby akumulację uszkodzeń DNA. Takie nadmierne uszkodzenie DNA często prowadzi do apoptozy. Jednak dodatkowy defekt w PMS2 może hamować tę apoptozę. wybrany dodatkowy niedobór PMS2 w obliczu zwiększonych uszkodzeń DNA, gdy występuje niedobór ERCC1 i/lub ERCC4 (XPF). Kiedy komórki jajnika chomika chińskiego z niedoborem ERCC1 były wielokrotnie poddawane uszkodzeniom DNA, z pięciu klonów pochodzących z komórek, które przeżyły, trzy zostały zmutowane w Pms2.
Progresja do raka okrężnicy
Podwójnie zmutowane komórki jajnika chomika chińskiego ERCC1, PMS2 po wystawieniu na działanie światła ultrafioletowego (czynnik uszkadzający DNA) wykazały 7375-krotnie większą częstotliwość mutacji niż komórki jajnika chomika chińskiego typu dzikiego i 967-krotnie większą częstotliwość mutacji niż komórki wadliwe w ERCC1, sam. Zatem niedobór komórek okrężnicy zarówno w ERCC1, jak i PMS2 powoduje niestabilność genomu . Podobna genetycznie niestabilna sytuacja spodziewana jest w przypadku komórek podwójnie defektywnych pod względem PMS2 i ERCC4 (XPF). Ta niestabilność prawdopodobnie zwiększyłaby progresję do raka okrężnicy, powodując fenotyp mutatora i odpowiada za obecność komórek z podwójnym niedoborem PMS2 i ERCC1 [lub PMS2 i ERCC4 (XPF)] w defektach terenowych związanych z rakiem okrężnicy. Jak wskazali Harper i Elledge, defekty w zdolności do prawidłowej odpowiedzi na uszkodzenia DNA i ich naprawy leżą u podstaw wielu form raka.
Linki zewnętrzne
- Często zadawane pytania dotyczące HNPCC zarchiwizowane 2007-08-15 w Wayback Machine z Narodowego Instytutu Zdrowia
- Wpis GeneReviews/NCBI/NIH/UW dotyczący zespołu Lyncha
|
Galeria WP
| |
|---|---|
|


