Biokoniugacja
Biokoniugacja to strategia chemiczna mająca na celu utworzenie stabilnego wiązania kowalencyjnego między dwiema cząsteczkami, z których przynajmniej jedna jest biomolekułą .
Funkcjonować
Ostatnie postępy w zrozumieniu biomolekuł umożliwiły ich zastosowanie w wielu dziedzinach, takich jak medycyna i materiały. Syntetycznie modyfikowane biomolekuły mogą mieć różnorodne funkcje, takie jak śledzenie zdarzeń komórkowych, ujawnianie funkcji enzymów , określanie biodystrybucji białek , obrazowanie specyficznych biomarkerów i dostarczanie leków do docelowych komórek. Biokoniugacja to kluczowa strategia, która łączy te zmodyfikowane biomolekuły z różnymi substratami .
Synteza
Synteza biokoniugatów wiąże się z różnymi wyzwaniami, począwszy od prostego i nieswoistego użycia barwnika fluorescencyjnego do złożonego projektowania koniugatów przeciwciało-lek . W rezultacie opracowano różne reakcje biokoniugacji – reakcje chemiczne łączące ze sobą dwie biomolekuły – w celu chemicznej modyfikacji białek. Typowe typy reakcji biokoniugacji na białkach to sprzęganie reszt aminokwasowych lizyny , cysteiny i tyrozyny ; oraz modyfikacja reszt aminokwasowych tryptofanu oraz N- i C-końca .
Jednak tym reakcjom często brakuje chemoselektywności i wydajności, ponieważ zależą one od obecności natywnych reszt aminokwasowych, które są zwykle obecne w dużych ilościach, które utrudniają selektywność. Istnieje rosnące zapotrzebowanie na strategie chemiczne, które mogą skutecznie przyłączać syntetyczne cząsteczki do białek. Jedną ze strategii jest najpierw zainstalowanie unikalnej grupy funkcyjnej na białku, a następnie reakcja bioortogonalna lub typu klik jest stosowana do połączenia biocząsteczki z tą unikalną grupą funkcyjną. Reakcje bioortogonalne ukierunkowane na nienatywne grupy funkcyjne są szeroko stosowane w chemii biokoniugacji. Niektóre ważne reakcje to modyfikacja ketonów i aldehydów , ligacja Staudingera z organicznymi azydkami , katalizowana miedzią cykloaddycja azydków Huisgena i cykloaddycja azydków Huisgena pod wpływem szczepu.
Typowe reakcje biokoniugacji
Najczęstsze biokoniugacje to sprzęganie małej cząsteczki (takiej jak biotyna lub barwnik fluorescencyjny) z białkiem lub koniugacje białko-białko, takie jak sprzęganie przeciwciała z enzymem. Inne mniej powszechne cząsteczki stosowane w biokoniugacji to oligosacharydy , kwasy nukleinowe , polimery syntetyczne, takie jak glikol polietylenowy i nanorurki węglowe . Koniugaty przeciwciało-lek, takie jak brentuksymab vedotin i gemtuzumab ozogamycyny , są również przykładami biokoniugacji i stanowią aktywny obszar badań w przemyśle farmaceutycznym. Ostatnio biokoniugacja zyskała również na znaczeniu w nanotechnologicznych , takich jak biokoniugowane kropki kwantowe .
Reakcje reszt lizyny
Nukleofilowa reszta lizyny jest często celem biokoniugacji białek, zazwyczaj poprzez aminoreaktywne estry N - (NHS) hydroksysukcynoimidylowe . Aby uzyskać optymalną liczbę deprotonowanych reszt lizyny, pH roztworu wodnego musi być niższe od pKa grupy amoniowej lizyny , które wynosi około 10,5, więc typowe pH reakcji wynosi około 8 i 9. Powszechnym odczynnikiem do sprzęgania Reakcją jest NHS-ester (pokazany w pierwszej reakcji poniżej na Figurze 1 ), który reaguje z nukleofilową lizyną poprzez mechanizm acylowania lizyny . Innymi podobnymi odczynnikami są izocyjaniany i izotiocyjaniany , które podlegają podobnemu mechanizmowi (pokazanemu w drugiej i trzeciej reakcji na rycinie 1 poniżej). Fluorki benzoilu (pokazane w ostatniej reakcji poniżej na Figurze 1 ), które umożliwiają modyfikację białek lizyną w łagodnych warunkach (niska temperatura, fizjologiczne pH ), zostały ostatnio zaproponowane jako alternatywa dla klasycznie stosowanych odczynników specyficznych dla lizyny.

Reakcje reszt cysteiny
Ponieważ wolna cysteina rzadko występuje na powierzchni białka, jest doskonałym wyborem do chemoselektywnej modyfikacji. W warunkach zasadowych reszty cysteiny zostaną zdeprotonowane, aby wytworzyć tiolanowy , który będzie reagował z miękkimi elektrofilami , takimi jak maleimidy i jodoacetamidy (pokazane w pierwszych dwóch reakcjach na rycinie 2 poniżej). W rezultacie powstaje wiązanie węgiel-siarka . Inna modyfikacja reszt cysteiny obejmuje tworzenie wiązania dwusiarczkowego (pokazane w trzeciej reakcji na Figurze 2 ). Zredukowane reszty cysteiny reagują z egzogennymi dwusiarczkami, tworząc nowe wiązanie dwusiarczkowe na białku. Do napędzania reakcji często stosuje się nadmiar dwusiarczków, takich jak 2-tiopirydon i 3-karboksy-4-nitrotiofenol. Wykazano, że alkiny z niedoborem elektronów reagują selektywnie z resztami cysteinowymi białek w obecności innych nukleofilowych reszt aminokwasowych. W zależności od podstawienia alkinu, reakcje te mogą prowadzić do rozszczepienia (gdy stosuje się pochodne alkinonu) lub stabilnych hydrolitycznie biokoniugatów (gdy stosuje się 3-arylopropionitryle ; ostatnia reakcja poniżej na Figurze 2 ).

Reakcje reszt tyrozynowych
Reszty tyrozyny są stosunkowo niereaktywne; dlatego nie były popularnymi celami biokoniugacji. Niedawny rozwój wykazał, że tyrozyna może być modyfikowana poprzez elektrofilowych podstawień aromatycznych (EAS) i jest selektywna dla aromatycznego węgla sąsiadującego z fenolową grupą hydroksylową . Staje się to szczególnie przydatne w przypadku, gdy reszty cysteiny nie mogą być celem. W szczególności diazonium skutecznie łączy się z resztami tyrozyny ( sól diazoniowa pokazana jako odczynnik w pierwszej reakcji na rysunku 3 poniżej), a podstawnik odciągający elektrony w pozycji 4 soli diazoniowej może skutecznie zwiększyć wydajność reakcji. Cykliczna pochodna diazodikarboksyamidu, taka jak 4-fenylo-1,2,4-triazolo-3,5-dion (PTAD), została opisana jako selektywna biokoniugacja reszt tyrozyny (druga reakcja na rycinie 3 poniżej). Opisano również, że trójskładnikowa reakcja typu Mannicha z aldehydami i anilinami (ostatnia reakcja na fig. 3 ) jest względnie selektywna względem tyrozyny w łagodnie zoptymalizowanych warunkach reakcji.

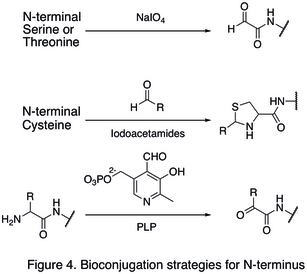
Reakcje N- i C-końca
Ponieważ naturalne reszty aminokwasowe są zwykle obecne w dużych ilościach, często trudno jest zmodyfikować jedno miejsce. Opracowano strategie ukierunkowane na końce białka, ponieważ znacznie zwiększyły one selektywność miejsca modyfikacji białka. Jedna z modyfikacji N-końca obejmuje funkcjonalizację końcowego aminokwasu. Utlenianie N-końcowych reszt seryny i treoniny może generować N-końcowy aldehyd, który może podlegać dalszym reakcjom bioortogonalnym (pokazanym w pierwszej reakcji na Figurze 4 ). Inny rodzaj modyfikacji obejmuje kondensację N-końcowej cysteiny z aldehydem, z wytworzeniem tiazolidyny , która jest stabilna przy wysokim pH (druga reakcja na Figurze 4 ). Stosując fosforan pirydoksalu (PLP), kilka N-końcowych aminokwasów można poddać transaminacji z wytworzeniem N-końcowego aldehydu , takiego jak glicyna i kwas asparaginowy (trzecia reakcja na Figurze 4 ).
Przykładem modyfikacji C-końca jest natywna ligacja chemiczna (NCL), która jest sprzęganiem C-końcowego tioestru i N-końcowej cysteiny ( Figura 5 ).

Reakcje bioortogonalne
Modyfikacja ketonów i aldehydów
Keton lub aldehyd można przyłączyć do białka poprzez utlenianie N-końcowych reszt seryny lub transaminację PLP. Dodatkowo można je wprowadzić przez włączenie nienaturalnych aminokwasów metodą Tirrella lub metodą Schultza. Następnie będą one selektywnie kondensować z alkoksyaminą i hydrazyną , tworząc pochodne oksymu i hydrazonu (pokazane odpowiednio w pierwszej i drugiej reakcji na Figurze 6 ). Ta reakcja jest wysoce chemoselektywna pod względem biokoniugacji białek, ale szybkość reakcji jest niska. Badania mechanistyczne pokazują, że etapem decydującym o szybkości jest odwodnienie tetraedrycznego związku pośredniego , dlatego często stosuje się łagodny roztwór kwaśny w celu przyspieszenia etapu odwodnienia.

Wprowadzenie katalizatora nukleofilowego może znacznie zwiększyć szybkość reakcji (pokazane na Figurze 7 ). Na przykład, stosując anilinę jako katalizator nukleofilowy, mniej zaludniony protonowany karbonyl staje się silnie zaludnionym protonowanym zasadą Schiffa . Innymi słowy, generuje wysokie stężenie reaktywnego elektrofilu. Ligacja oksymowa może wtedy zachodzić łatwo i doniesiono, że szybkość wzrastała do 400 razy w łagodnych warunkach kwasowych. Kluczem do tego katalizatora jest to, że może generować reaktywny elektrofil bez konkurowania z pożądanym produktem.

Niedawne odkrycia, które wykorzystują proksymalne grupy funkcyjne, umożliwiły działanie kondensacji hydrazonu przy 20 M -1 s -1 przy obojętnym pH, podczas gdy odkryto kondensacje oksymu, które zachodzą przy 500-10000 M -1 s -1 przy obojętnym pH bez dodanych katalizatorów.
Ligacja Staudigera z azydkami
Ligacja Staudingera azydków i fosfiny była szeroko stosowana w dziedzinie biologii chemicznej. Ponieważ jest w stanie tworzyć stabilne wiązanie amidowe w żywych komórkach i zwierzętach, zastosowano go do modyfikacji błony komórkowej , obrazowania in vivo i innych badań biokoniugacji.

W przeciwieństwie do klasycznej reakcji Staudingera, ligacja Staudingera jest reakcją drugiego rzędu, w której etapem ograniczającym szybkość jest tworzenie fosfazydu (specyficzny mechanizm reakcji pokazany na Figurze 9 ). Trifenylofosfina najpierw reaguje z azydkiem, dając azaylid przez czteroczłonowy stan przejściowy pierścienia, a następnie reakcja wewnątrzcząsteczkowa prowadzi do iminofosforanu jako związku pośredniego, który następnie daje wiązanie amidowe podczas hydrolizy.

Cyklizacja Huisgena azydków
Katalizowana miedzią cyklizacja azydków Huisgena
Azydek stał się popularnym celem chemoselektywnej modyfikacji białek, ponieważ są one małe i mają korzystny potencjał reakcji termodynamicznej . Jedną z takich reakcji azydkowych jest reakcja cykloaddycji [3+2] z alkinem , ale reakcja ta wymaga wysokiej temperatury i często daje mieszaniny regioizomerów .

Ulepszona reakcja opracowana przez chemika Karla Barry'ego Sharplessa obejmuje katalizator miedziowy (I), który sprzęga azydek z końcowym alkinem, dając tylko 1,4-podstawione 1,2,3 triazole z wysoką wydajnością (pokazane poniżej na Figurze 11 ). Badanie mechanistyczne sugeruje reakcję stopniową. Cu (I) najpierw łączy się z acetylenami , a następnie reaguje z azydkiem, tworząc sześcioczłonowy związek pośredni. Proces jest bardzo wytrzymały, ponieważ zachodzi przy pH w zakresie od 4 do 12, a siarczan miedzi (II) jest często stosowany jako katalizator w obecności środka redukującego .

Szczep promował cyklizację azydków Huisgena
Chociaż ligacja Staudingera jest odpowiednią biokoniugacją w żywych komórkach bez większej toksyczności, wrażliwość fosfiny na utlenianie powietrzem i jej słaba rozpuszczalność w wodzie znacznie utrudniają jej skuteczność. Katalizowane miedzią (I) sprzęganie azydek-alkin ma rozsądną szybkość reakcji i wydajność w warunkach fizjologicznych, ale miedź ma znaczną toksyczność i czasami zakłóca funkcje białek w żywych komórkach. W 2004 roku laboratorium chemika Carolyn R. Bertozzi opracowało cykloaddycję [3+2] pozbawioną metalu, używając naprężonego cyklooktynu i azydku. Cyklooktyn, który jest najmniejszym stabilnym cykloalkinem, może sprzęgać się z azydkiem poprzez [3+2] cykloaddycję, prowadząc do dwóch regioizomerycznych triazoli ( Figura 12 ). Reakcja zachodzi łatwo w temperaturze pokojowej i dlatego może być stosowana do skutecznej modyfikacji żywych komórek bez negatywnych skutków. Donoszono również, że instalacja fluorowych na cyklicznym alkinie może znacznie przyspieszyć szybkość reakcji.

Przykłady stosowanych technik biokoniugacji
Czynniki wzrostowe
Opisano biokoniugację TGF-β do nanocząstek tlenku żelaza i jego aktywację poprzez hipertermię magnetyczną in vitro. Dokonano tego, stosując 1-(3-dimetyloaminopropylo)etylokarbodiimid w połączeniu z N-hydroksysukcynoimidem w celu utworzenia pierwszorzędowych wiązań amidowych z wolnymi pierwszorzędowymi aminami na czynniku wzrostu. Nanorurki węglowe zostały z powodzeniem wykorzystane w połączeniu z biokoniugacją w celu połączenia TGF-β, a następnie aktywacji światłem bliskiej podczerwieni. Zazwyczaj reakcje te obejmowały użycie środka sieciującego, ale niektóre z nich zwiększają przestrzeń molekularną między związkiem będącym przedmiotem zainteresowania a materiałem podstawowym, co z kolei powoduje wyższy stopień niespecyficznego wiązania i niepożądanej reaktywności.
Zobacz też
- Immunofluorescencja
- Inżynieria biomolekularna
- Biotynylacja
- SpyTag/SpyCatcher
- Nienaturalne aminokwasy
- Czasopismo Bioconjugate Chemistry