Metakody kreskowe
| Część serii o |
| kodowaniu kreskowym DNA |
|---|

|
| Według taksonów |
| Inny |
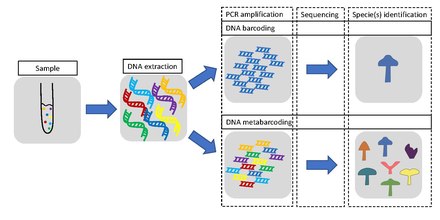
Metabarcoding to kod kreskowy DNA / RNA (lub eDNA / eRNA ) w sposób umożliwiający jednoczesną identyfikację wielu taksonów w obrębie tej samej próbki. Główna różnica między kodowaniem kreskowym a metakodowaniem kreskowym polega na tym, że metakodowanie kreskowe nie koncentruje się na jednym konkretnym organizmie, ale ma na celu określenie składu gatunkowego w próbce.
Kod kreskowy składa się z krótkiego zmiennego regionu genu (na przykład patrz różne markery/kody kreskowe ), który jest przydatny do przypisania taksonomicznego, otoczony przez wysoce konserwatywne regiony genów, które można wykorzystać do zaprojektowania startera . Ten pomysł ogólnego kodowania kreskowego zrodził się w 2003 roku od naukowców z University of Guelph .
Procedura metakodowania kreskowego, podobnie jak ogólne kodowanie kreskowe, przebiega kolejno przez etapy ekstrakcji DNA , amplifikacji PCR , sekwencjonowania i analizy danych . W zależności od tego, czy celem jest kod kreskowy pojedynczego gatunku, czy też metakodowanie kilku gatunków, stosuje się różne geny. W tym drugim przypadku stosuje się bardziej uniwersalny gen. Metabarcoding nie wykorzystuje DNA/RNA pojedynczego gatunku jako punktu wyjścia, ale DNA/RNA z kilku różnych organizmów pochodzących z jednej próbki środowiskowej lub zbiorczej.
Środowiskowe DNA
Środowiskowy DNA lub eDNA opisuje materiał genetyczny obecny w próbkach środowiskowych, takich jak osad, woda i powietrze, w tym całe komórki, pozakomórkowe DNA i potencjalnie całe organizmy. eDNA można przechwytywać z próbek środowiskowych i konserwować , ekstrahować , amplifikować , sekwencjonować i kategoryzować na podstawie jego sekwencji. Na podstawie tych informacji możliwe jest wykrywanie i klasyfikacja gatunków. eDNA może pochodzić ze skóry, śluzu, śliny, nasienia, wydzielin, jaj, kału, moczu, krwi, korzeni, liści, owoców, pyłków i gnijących ciał większych organizmów, podczas gdy mikroorganizmy można uzyskać w całości. Produkcja eDNA zależy od biomasy , wieku i aktywności żywieniowej organizmu, a także fizjologii, historii życia i wykorzystania przestrzeni.
Do 2019 roku metody badań eDNA zostały rozszerzone, aby móc oceniać całe społeczności na podstawie jednej próbki. Proces ten obejmuje metabarkodowanie, które można precyzyjnie zdefiniować jako użycie ogólnych lub uniwersalnych starterów reakcji łańcuchowej polimerazy (PCR) na mieszanych próbkach DNA dowolnego pochodzenia, a następnie wysokoprzepustowe sekwencjonowanie nowej generacji (NGS) w celu określenia składu gatunkowego próbki. Metoda ta jest powszechna w mikrobiologii od lat, ale od 2020 roku dopiero znajduje zastosowanie w ocenie makroorganizmów. Zastosowania metabarkodów eDNA w całym ekosystemie mogą nie tylko opisywać społeczności i różnorodność biologiczną, ale także wykrywać interakcje i ekologię funkcjonalną w dużych skalach przestrzennych, chociaż może to być ograniczone przez fałszywe odczyty spowodowane zanieczyszczeniem lub innymi błędami. Ogólnie rzecz biorąc, metabarkodowanie eDNA zwiększa szybkość, dokładność i identyfikację w porównaniu z tradycyjnymi kodami kreskowymi i zmniejsza koszty, ale musi być ustandaryzowane i ujednolicone, integrując taksonomię i metody molekularne w celu przeprowadzenia pełnych badań ekologicznych.


Metabarcoding eDNA ma zastosowanie do monitorowania różnorodności we wszystkich siedliskach i grupach taksonomicznych, rekonstrukcji starożytnych ekosystemów, interakcji między roślinami a zapylaczami, analizie diety, wykrywaniu gatunków inwazyjnych, reakcjach na zanieczyszczenia i monitorowaniu jakości powietrza. Metabarkodowanie eDNA to unikalna metoda, która wciąż jest w fazie rozwoju i prawdopodobnie przez jakiś czas będzie się zmieniać wraz z postępem technologicznym i standaryzacją procedur. Jednak w miarę optymalizacji kodów kreskowych i upowszechniania się ich stosowania prawdopodobnie stanie się niezbędnym narzędziem do monitorowania ekologicznego i globalnych badań konserwatorskich.
DNA społeczności
Od początku sekwencjonowania o dużej przepustowości ( HTS ) wykorzystanie metabarkodów jako narzędzia do wykrywania różnorodności biologicznej wzbudziło ogromne zainteresowanie. Jednak nie ma jeszcze jasności co do tego, jaki materiał źródłowy jest używany do przeprowadzania analiz metabarkodów (np. DNA środowiska a DNA społeczności ). Bez jasności między tymi dwoma materiałami źródłowymi różnice w pobieraniu próbek, a także różnice w procedurach laboratoryjnych mogą wpływać na kolejne potoki bioinformatyczne wykorzystywane do przetwarzania danych i komplikować interpretację przestrzennych i czasowych wzorców różnorodności biologicznej. W tym przypadku staramy się wyraźnie rozróżnić dominujące materiały źródłowe i ich wpływ na dalszą analizę i interpretację metabarkodowania środowiskowego DNA zwierząt i roślin w porównaniu z metabarkodowaniem DNA społeczności.
W przypadku metabarkodowania wspólnotowego DNA zwierząt i roślin grupy docelowe są najczęściej zbierane masowo (np. gleba, pułapka na złe samopoczucie lub sieć), a osobniki są usuwane z innych szczątków próbek i łączone razem przed masową ekstrakcją DNA. Natomiast eDNA makroorganizmów jest izolowane bezpośrednio z materiału środowiskowego (np. gleby lub wody) bez uprzedniej segregacji poszczególnych organizmów lub materiału roślinnego z próbki i domyślnie zakłada, że cały organizm nie jest obecny w próbce. Oczywiście próbki DNA społeczności mogą zawierać DNA z części tkanek, komórek i organelli innych organizmów (np. zawartość jelit, DNA wewnątrzkomórkowe lub zewnątrzkomórkowe skóry). Podobnie, próbki eDNA makroorganizmów mogą przypadkowo uchwycić całe mikroskopijne organizmy inne niż docelowe (np. protisty, bakterie). Zatem rozróżnienie to może przynajmniej częściowo załamać się w praktyce.
Inną ważną różnicą między DNA społeczności a eDNA makroorganizmów jest to, że sekwencje wygenerowane z metabarkodów DNA społeczności można zweryfikować taksonomicznie, gdy okazy nie zostaną zniszczone w procesie ekstrakcji. W tym przypadku sekwencje można następnie wygenerować z próbek kuponów przy użyciu sekwencjonowania Sangera. Ponieważ próbki do metabarkodowania eDNA nie zawierają całych organizmów, nie można przeprowadzić takich porównań in situ. Powinowactwa taksonomiczne można zatem ustalić jedynie poprzez bezpośrednie porównanie uzyskanych sekwencji (lub poprzez wygenerowane bioinformatycznie operacyjne jednostki taksonomiczne (MOTU)) z sekwencjami, które są taksonomicznie opatrzone adnotacjami, takimi jak baza danych nukleotydów GenBank NCBI, BOLD lub do samodzielnie wygenerowanych referencyjnych baz danych z DNA zsekwencjonowanego metodą Sangera. ( Operacyjna jednostka taksonomiczna molekularna (MOTU) to grupa identyfikowana za pomocą algorytmów klastrowych i z góry określonego procentowego podobieństwa sekwencji, na przykład 97%). Następnie, aby przynajmniej częściowo potwierdzić powstałą listę taksonów, dokonuje się porównań z konwencjonalnymi metodami badań fizycznych, akustycznych lub wizualnych prowadzonymi w tym samym czasie lub porównuje się je z historycznymi zapisami z badań dla danej lokalizacji (patrz Tabela 1).
Różnica w materiale źródłowym między DNA społeczności a eDNA ma zatem wyraźne konsekwencje dla interpretacji skali wnioskowania dla czasu i przestrzeni na temat wykrytej różnorodności biologicznej. Z DNA społeczności jasno wynika, że poszczególne gatunki zostały znalezione w tym czasie i miejscu, ale w przypadku eDNA organizm, który wytworzył DNA, może znajdować się powyżej miejsca, z którego pobrano próbkę, lub DNA mogło zostać przetransportowane w odchodach bardziej mobilne gatunki drapieżne (np. ptaki deponujące eDNA ryb lub były wcześniej obecne, ale nie są już aktywne w społeczności, a wykrywanie odbywa się na podstawie DNA, które zostało wyrzucone lata lub dekady wcześniej. To ostatnie oznacza, że skala wnioskowania zarówno w przestrzeni, jak i w czasie należy dokładnie rozważyć przy wnioskowaniu o obecności gatunku w zbiorowisku na podstawie eDNA.
Etapy metakodowania kreskowego

Istnieje sześć etapów lub etapów kodowania kreskowego i metabarkodowania DNA. Kody kreskowe DNA zwierząt (a konkretnie nietoperzy ) są używane jako przykład na diagramie po prawej stronie oraz w dyskusji bezpośrednio poniżej.
Najpierw wybiera się odpowiednie regiony kodu kreskowego DNA, aby odpowiedzieć na określone pytanie badawcze. Najczęściej używanym regionem kodu kreskowego DNA dla zwierząt jest segment o długości około 600 par zasad mitochondrialnego genu oksydazy cytochromowej I (CO1). To locus zapewnia dużą zmienność sekwencji między gatunkami, ale stosunkowo niewielką zmienność w obrębie gatunku. Innymi powszechnie używanymi regionami kodu kreskowego używanymi do identyfikacji gatunków zwierząt są rybosomalnego DNA (rDNA), takie jak 16S , 18S i 12S oraz regiony mitochondrialne, takie jak cytochrom B. Te znaczniki mają zalety i wady i są używane do różnych celów. Dłuższe regiony kodu kreskowego (o długości co najmniej 600 par zasad) są często potrzebne do dokładnego rozgraniczenia gatunków, zwłaszcza do rozróżnienia bliskich krewnych. Identyfikacja producenta szczątków organizmów, takich jak odchody, włosy i ślina, może służyć jako środek zastępczy w celu zweryfikowania braku/obecności gatunku w ekosystemie. DNA w tych szczątkach jest zwykle niskiej jakości i ilości, dlatego w takich przypadkach stosuje się krótsze kody kreskowe o długości około 100 par zasad. Podobnie DNA pozostające w łajnie również często ulega degradacji, więc do identyfikacji zjedzonej ofiary potrzebne są krótkie kody kreskowe.
Po drugie, należy zbudować referencyjną bazę danych zawierającą wszystkie kody kreskowe DNA, które mogą wystąpić w badaniu. Idealnie byłoby, gdyby te kody kreskowe były generowane z okazów objętych kuponami zdeponowanych w publicznie dostępnym miejscu, takim jak na przykład muzeum historii naturalnej lub inny instytut badawczy. Tworzenie takich referencyjnych baz danych odbywa się obecnie na całym świecie. Organizacje partnerskie współpracują w międzynarodowych projektach, takich jak International Barcode of Life Project (iBOL) i Consortium for the Barcode of Life (CBOL), którego celem jest skonstruowanie odniesienia do kodu kreskowego DNA, który będzie podstawą do identyfikacji światowego biomu na podstawie DNA. Dobrze znane repozytoria kodów kreskowych to NCBI GenBank i Barcode of Life Data System (BOLD).
Po trzecie, komórki zawierające DNA będące przedmiotem zainteresowania muszą zostać otwarte, aby odsłonić jego DNA. Ten etap, i oczyszczanie DNA , należy przeprowadzić z badanego substratu. W tym celu dostępnych jest kilka procedur. Należy wybrać specjalne techniki w celu wyizolowania DNA z substratów z częściowo zdegradowanym DNA, na przykład próbek kopalnych i próbek zawierających inhibitory, takich jak krew, kał i gleba. Ekstrakcje, w przypadku których oczekuje się niskiej wydajności lub jakości DNA, należy przeprowadzać na starożytnym DNA placówki, wraz z ustalonymi protokołami, aby uniknąć zanieczyszczenia współczesnym DNA. Eksperymenty należy zawsze przeprowadzać w dwóch powtórzeniach z uwzględnieniem kontroli pozytywnych.
Po czwarte, amplikony muszą być generowane z DNA wyekstrahowanego z pojedynczej próbki lub ze złożonych mieszanin starterów opartych na kodach kreskowych DNA wybranych w kroku 1. Aby śledzić ich pochodzenie, znakowane nukleotydy (identyfikatory molekularne lub znaczniki MID) muszą być dodawane w przypadku metabarkodów. Etykiety te są potrzebne później w analizach, aby śledzić odczyty z zestawu danych zbiorczych z powrotem do ich pochodzenia.

Po piąte, należy wybrać odpowiednie techniki sekwencjonowania DNA . Klasyczna metoda terminacji łańcucha Sangera polega na selektywnym włączaniu wydłużających łańcuch inhibitorów polimerazy DNA podczas replikacji DNA . Te cztery zasady rozdziela się według wielkości za pomocą elektroforezy , a następnie identyfikuje za pomocą detekcji laserowej. Metoda Sangera jest ograniczona i może generować pojedynczy odczyt w tym samym czasie, dlatego nadaje się do generowania kodów kreskowych DNA z substratów zawierających tylko jeden gatunek. Nowe technologie, takie jak sekwencjonowanie nanoporów spowodowały obniżenie kosztu sekwencjonowania DNA z około 30 000 USD za megabajt w 2002 r. do około 0,60 USD w 2016 r. Nowoczesne technologie sekwencjonowania nowej generacji (NGS) mogą obsłużyć od tysięcy do milionów odczytów równolegle i dlatego nadają się do masowej identyfikacji mieszanka różnych gatunków obecnych w podłożu, podsumowana jako metakod kreskowy.
Wreszcie, należy przeprowadzić analizy bioinformatyczne , aby dopasować kody kreskowe DNA uzyskane z numerami indeksów kodów kreskowych (BIN) w bibliotekach referencyjnych. Każdy BIN lub klaster BIN można zidentyfikować na poziomie gatunku, jeśli wykazuje wysoką (>97%) zgodność z kodami kreskowymi DNA powiązanymi z gatunkiem występującym w bibliotece referencyjnej lub gdy nadal brakuje identyfikacji taksonomicznej na poziomie gatunku , jednostka taksonomiczna (OTU), która odnosi się do grupy gatunków (tj. rodzaju, rodziny lub wyższej rangi taksonomicznej). (Patrz sortowanie (metagenomika) ). Wyniki rurociągu bioinformatycznego muszą zostać przycięte, na przykład poprzez odfiltrowanie niewiarygodnych singletonów, zbędnych duplikatów, odczytów niskiej jakości i/lub odczytów chimerycznych . Na ogół odbywa się to poprzez przeprowadzanie seryjnych wyszukiwań BLAST w połączeniu z automatycznym filtrowaniem i przycinaniem skryptów. Standaryzowane progi są potrzebne do rozróżnienia między różnymi gatunkami lub do prawidłowej i błędnej identyfikacji.
Przepływ pracy z kodami kreskowymi
Pomimo oczywistej siły tego podejścia, metabarkodowanie eDNA podlega wyzwaniom związanym z precyzją i dokładnością występującymi w całym przepływie pracy w terenie, w laboratorium i przy klawiaturze. Jak pokazano na diagramie po prawej stronie, po wstępnym projekcie badania (hipoteza/pytanie, docelowa grupa taksonomiczna itp.) obecny przepływ pracy eDNA składa się z trzech elementów: terenowego, laboratoryjnego i bioinformatycznego. Komponent terenowy składa się z pobierania próbek (np. wody, osadu, powietrza), które są konserwowane lub zamrażane przed ekstrakcją DNA. Element laboratoryjny składa się z czterech podstawowych etapów: (i) DNA jest zagęszczane (jeśli nie jest przeprowadzane w terenie) i oczyszczane, (ii) PCR stosuje się do amplifikacji docelowego genu lub regionu, (iii) unikalne sekwencje nukleotydowe zwane „indeksami” (określane również jako „kody kreskowe”) są włączane za pomocą PCR lub są łączone (wiązane) z różnymi produktami PCR, tworząc „ bibliotekę ” dzięki czemu wiele próbek można połączyć razem, a (iv) połączone biblioteki są następnie sekwencjonowane na maszynie o wysokiej przepustowości . Ostatnim krokiem po laboratoryjnym przetwarzaniu próbek jest przetwarzanie obliczeniowe plików wyjściowych z sekwensera przy użyciu solidnego potoku bioinformatycznego.


OTU i pojęcie gatunku
Metoda i wizualizacja

a) Różnorodność alfa wyświetlana jako wykresy słupkowe taksonomii, pokazujące względną obfitość taksonów w próbkach przy użyciu struktury wizualizacji danych Phinch (Bik & Pitch Interactive 2014). b) Wzory różnorodności beta zilustrowane za pomocą analiz głównych współrzędnych przeprowadzonych w QIIME , gdzie każda kropka reprezentuje próbkę, a kolory rozróżniają różne klasy próbek. Im bliżej dwa punkty próbkowania w przestrzeni 3D, tym bardziej podobne są ich zbiorowiska c) GraPhalAn filogenetyczna wizualizacja danych środowiskowych, z okrągłymi mapami cieplnymi
oraz słupki obfitości używane do przekazywania ilościowych cech taksonów. d) Edge PCA, oparta na drzewie metryka różnorodności, która identyfikuje określone linie (zielone / pomarańczowe gałęzie), które najbardziej przyczyniają się do zmian społeczności obserwowanych w próbkach rozmieszczonych na różnych osiach PCA.
Metoda wymaga zarchiwizowania każdego zebranego DNA wraz z odpowiadającym mu „okazem typu” (po jednym dla każdego taksonu), oprócz zwykłych danych zbierania. Typy te są przechowywane w określonych instytucjach (muzea, laboratoria molekularne, uniwersytety, ogrody zoologiczne, ogrody botaniczne, zielniki itp.) po jednym dla każdego kraju, aw niektórych przypadkach ta sama instytucja jest przypisana do przechowywania typów z więcej niż jednego kraju , w przypadkach, gdy niektóre narody nie mają technologii lub środków finansowych, aby to zrobić.
W ten sposób tworzenie typowych okazów kodów genetycznych reprezentuje metodologię równoległą do tej przeprowadzanej przez tradycyjną taksonomię.
W pierwszym etapie zdefiniowano region DNA, który zostanie użyty do stworzenia kodu kreskowego. Musiał być krótki i osiągnąć wysoki procent unikalnych sekwencji. W przypadku zwierząt, alg i grzybów część genu mitochondrialnego, która koduje podjednostkę 1 enzymu oksydazy cytochromowej, CO1, zapewnia wysoki odsetek (95%), region około 648 par zasad.
W przypadku roślin użycie CO1 nie było skuteczne, ponieważ mają one niski poziom zmienności w tym regionie, oprócz trudności, które są spowodowane częstymi efektami poliploidii, introgresji i hybrydyzacji , więc genom chloroplastów wydaje się bardziej odpowiedni .
Aplikacje
Sieci zapylaczy

↑ kod kreskowy ↑ ankiety wizytacyjne (a,b) grupy roślin-zapylaczy (c,d) gatunki roślin-zapylaczy (e,f) poszczególne gatunki roślin-zapylaczy ( Empis leptempis pandellei )
Apis: Apis mellifera ; Bomba: Bombus sp.; W.bee: dzikie pszczoły; O.Hym.: inne błonkoskrzydłe ; O.Dipt.: Inne Diptera ; Emp.: Empididae ; Syrph.: Syrphidae ; płk: Coleoptera ; Lep.: Lepidoptera ; Musc.: Muscidae . Grubość linii podkreśla proporcje interakcji
Diagram po prawej stronie pokazuje porównanie sieci zapylania opartych na metabarkodzie DNA z bardziej tradycyjnymi sieciami opartymi na bezpośrednich obserwacjach wizyt owadów na roślinach. Wykrywając liczne dodatkowe ukryte interakcje, dane metakodów kreskowych w dużym stopniu zmieniają właściwości sieci zapylania w porównaniu z badaniami wizyt. Dane molekularne pokazują, że owady zapylające są znacznie bardziej ogólne , niż można by się spodziewać na podstawie ankiet wizytacyjnych. Jednak gatunki zapylaczy składały się ze stosunkowo wyspecjalizowanych osobników i tworzyły grupy funkcjonalne wysoce wyspecjalizowane w odmianach kwiatowych .
W konsekwencji zachodzących globalnych zmian dramatyczny i równoległy światowy spadek liczby owadów zapylających zaobserwowano gatunki roślin zapylanych przez zwierzęta. Zrozumienie reakcji sieci zapylania na te spadki jest pilnie potrzebne do zdiagnozowania zagrożeń, jakie mogą ponosić ekosystemy, a także do zaprojektowania i oceny skuteczności działań ochronnych. Wczesne badania nad zapylaniem zwierząt dotyczyły systemów uproszczonych, tj. specyficznych interakcji parami lub małych podzbiorów zbiorowisk roślinno-zwierzęcych. Jednak wpływ zakłóceń zachodzi poprzez wysoce złożone sieci interakcji, a obecnie te złożone systemy są obecnie głównym przedmiotem badań. Ocena prawdziwych sieci (określonych w procesie ekologicznym) na podstawie badań terenowych, które podlegają efektom pobierania próbek, nadal stanowi wyzwanie.
Ostatnie badania naukowe wyraźnie skorzystały z koncepcji i narzędzi sieciowych do badania wzorców interakcji w dużych zbiorowiskach gatunków. Wykazali, że sieci roślin-zapylaczy mają wysoce ustrukturyzowaną strukturę, znacznie odbiegającą od przypadkowych powiązań. Zwykle sieci mają (1) niski poziom powiązań (zrealizowany ułamek wszystkich potencjalnych powiązań w społeczności), co sugeruje niski stopień uogólnienia; (2) wysokie zagnieżdżenie (im bardziej wyspecjalizowane organizmy są bardziej skłonne do interakcji z podzbiorami gatunków, z którymi wchodzą w interakcje organizmy bardziej ogólne), bardziej wyspecjalizowane gatunki wchodzą w interakcje tylko z odpowiednimi podzbiorami tych gatunków wchodzących w interakcje z bardziej ogólnymi; (3) skumulowany rozkład połączeń (liczba połączeń na gatunek, s), który następuje po potędze lub okrojonej funkcji prawa potęgowego, charakteryzujący się kilkoma supergeneralistami z większą liczbą połączeń niż oczekiwano przez przypadek i wieloma specjalistami; (4) organizacja modułowa. Moduł to grupa gatunków roślin i zapylaczy, która wykazuje wysoki poziom łączności wewnątrz modułu i która jest słabo powiązana z gatunkami innych grup.
Niski poziom łączności i wysoki odsetek specjalistów zajmujących się sieciami zapylania kontrastują z poglądem, że w sieciach normą jest uogólnienie, a nie specjalizacja. Rzeczywiście, większość gatunków roślin jest odwiedzana przez różnorodne owady zapylające, które wykorzystują zasoby kwiatowe z szerokiej gamy gatunków roślin. Główną przyczyną przywoływaną w celu wyjaśnienia tej pozornej sprzeczności jest niepełne próbkowanie interakcji. Rzeczywiście, większość właściwości sieci jest bardzo wrażliwa na intensywność próbkowania i rozmiar sieci. Badania sieci są zasadniczo fitocentryczne, tj. oparte na obserwacjach wizyt owadów zapylających na kwiatach. To skoncentrowane na roślinach podejście ma jednak nieodłączne ograniczenia, które mogą utrudniać zrozumienie mechanizmów przyczyniających się do gromadzenia się społeczności i wzorców różnorodności biologicznej. Po pierwsze, bezpośrednie obserwacje wizyt owadów zapylających u niektórych taksonów, takich jak storczyki, są często rzadkie, a rzadkie interakcje są ogólnie bardzo trudne do wykrycia w terenie. Zbiorowiska owadów zapylających i roślin zwykle składają się z niewielu gatunków występujących w obfitości i wielu gatunków rzadkich, które są słabo rejestrowane w badaniach wizytacyjnych. Te rzadkie gatunki pojawiają się jako specjaliści, podczas gdy w rzeczywistości mogą być typowymi generalistami. Ze względu na pozytywny związek między częstotliwością interakcji (f) a łącznością (s), niedopróbkowane interakcje mogą prowadzić do przeszacowania stopnia specjalizacji sieci. Po drugie, analizy sieci działały głównie na poziomie gatunków. Sieci bardzo rzadko były skalowane w górę do grup funkcjonalnych lub w dół do sieci opartych na jednostkach, a większość z nich skupiała się tylko na jednym lub dwóch gatunkach. Zachowanie osobników lub kolonii jest powszechnie ignorowane, chociaż może wpływać na strukturę sieci gatunkowych. Gatunki uznawane za ogólne w sieciach gatunków mogą zatem pociągać za sobą tajemnicze wyspecjalizowane osobniki lub kolonie. Po trzecie, odwiedzający kwiaty nie zawsze są skutecznymi zapylaczami, ponieważ mogą nie składać pyłku tego samego gatunku i/lub dużo pyłku heterospecyficznego. Podejścia skoncentrowane na zwierzętach, oparte na badaniu obciążenia pyłkami gości i znamionach roślin, mogą być bardziej skuteczne w ujawnianiu interakcji między roślinami a zapylaczami.
Rozplątywanie sieci pokarmowych

Metabarcoding oferuje nowe możliwości rozszyfrowania powiązań troficznych między drapieżnikami a ich ofiarami w sieciach pokarmowych. W porównaniu z tradycyjnymi, czasochłonnymi metodami, takimi jak analizy mikroskopowe czy serologiczne , rozwój metabarkodowania DNA umożliwia identyfikację gatunków ofiar bez wcześniejszej wiedzy o zasięgu ofiar drapieżników. Ponadto metabarkodowania można również użyć do scharakteryzowania dużej liczby gatunków w jednym PCR reakcji i analizować kilkaset próbek jednocześnie. Takie podejście jest coraz częściej wykorzystywane do badania różnorodności funkcjonalnej i struktury sieci pokarmowych w agroekosystemach. Podobnie jak inne podejścia molekularne, metabarkodowanie daje jedynie wyniki jakościowe dotyczące obecności/nieobecności gatunków ofiar w próbkach jelita lub kału. Jednak ta wiedza o tożsamości zdobyczy konsumowanej przez drapieżniki tego samego gatunku w danym środowisku umożliwia „pragmatyczny i użyteczny surogat prawdziwie ilościowych informacji.
W ekologii sieci troficznej „kto zjada kogo” jest fundamentalną kwestią dla lepszego zrozumienia złożonych interakcji troficznych istniejących między szkodnikami a ich naturalnymi wrogami w danym ekosystemie. Analiza dietetyczna drapieżników stawonogów i kręgowców umożliwia identyfikację kluczowych drapieżników zaangażowanych w naturalną kontrolę szkodników stawonogów i daje wgląd w zakres ich diety ( ogólnie vs. specjalista ) oraz drapieżnictwo wewnątrz cechu .
Diagram po prawej stronie podsumowuje wyniki badania przeprowadzonego w 2020 r., w którym wykorzystano kody kreskowe do rozwikłania funkcjonalnej różnorodności i struktury sieci pokarmowej związanej z kilkoma polami prosa w Senegalu. Po przypisaniu zidentyfikowanych OTU jako gatunków zidentyfikowano 27 taksonów ofiar stawonogów spośród dziewięciu drapieżników stawonogów. Średnia liczba taksonów ofiar wykrytych w próbce była najwyższa u chrząszczy biegaczowatych , mrówek i pająków, a najniższa u pozostałych drapieżników, w tym pluskwiaków , pentatomidów pluskwy i skorki. Wśród drapieżnych stawonogów zaobserwowano dużą różnorodność ofiar stawonogów u pająków, chrząszczy biegaczowatych, mrówek i antokorydów. Z kolei różnorodność gatunków zdobyczy zidentyfikowanych w skorkach i robakach pentatomidowych była stosunkowo niewielka. Lepidoptera , Hemiptera , Diptera i Coleoptera były najczęstszymi taksonami ofiar owadów wykrytymi u drapieżnych stawonogów.
Zachowanie funkcjonalnej różnorodności biologicznej i powiązanych usług ekosystemowych , zwłaszcza poprzez zwalczanie szkodników przy użyciu ich naturalnych wrogów, oferuje nowe możliwości sprostania wyzwaniom związanym ze zrównoważoną intensyfikacją systemów produkcji żywności. Drapieżnictwo szkodników upraw przez drapieżniki ogólne, w tym stawonogi i kręgowce, jest głównym elementem naturalnego zwalczania szkodników . Szczególnie ważną cechą większości drapieżników ogólnych jest to, że mogą kolonizować uprawy na początku sezonu, najpierw żerując na alternatywnej zdobyczy. Jednak zakres diety „ogólnej” pociąga za sobą pewne wady zwalczania szkodników, takie jak drapieżnictwo wewnątrz gildii. Dostrojona diagnoza szerokości diety drapieżników ogólnych, w tym drapieżnictwa ofiar innych niż szkodniki, jest zatem potrzebna, aby lepiej rozplątać sieci pokarmowe (np. konkurencja eksploatacyjna i pozorna konkurencja) i ostatecznie zidentyfikować kluczowe czynniki napędzające naturalną kontrolę szkodników w agroekosystemach. Jednak znaczenie ogólnych drapieżników w sieci troficznej jest generalnie trudne do oszacowania ze względu na efemeryczny charakter indywidualnych interakcji drapieżnik-ofiara. Jedynym rozstrzygającym dowodem na drapieżnictwo jest bezpośrednia obserwacja spożycia ofiary, identyfikacja pozostałości ofiary w jelitach drapieżnika oraz analiza treści pokarmowej lub odchodów.
Bioasekuracja morska

.jpeg)


Rozprzestrzenianie się gatunków nierodzimych (NIS) stanowi znaczące i rosnące zagrożenie dla ekosystemów. W systemach morskich NIS, które przetrwają transport i dostosują się do nowych lokalizacji, mogą mieć znaczący niekorzystny wpływ na lokalną różnorodność biologiczną, w tym przemieszczenie gatunków rodzimych oraz zmiany w społecznościach biologicznych i związanych z nimi sieciach pokarmowych. Po ustanowieniu NIS ich wyeliminowanie jest niezwykle trudne i kosztowne, a dalsze rozprzestrzenianie się regionalne może nastąpić poprzez naturalne rozprzestrzenianie się lub poprzez antropogeniczne szlaki transportowe. Podczas gdy zanieczyszczenie kadłubów statków i wody balastowe statków są dobrze znane jako ważne antropogeniczne drogi rozprzestrzeniania się NIS na skalę międzynarodową, stosunkowo niewiele wiadomo o potencjale regionalnych statków tranzytowych w zakresie przyczyniania się do wtórnego rozprzestrzeniania się szkodników morskich poprzez przemieszczanie wody zęzowej.
Niedawne badania wykazały, że woda i związane z nią zanieczyszczenia unoszone w przestrzeniach zęzowych małych statków (<20 m) mogą działać jako wektor rozprzestrzeniania się NIS na skalę regionalną. Woda zęzowa jest definiowana jako jakakolwiek woda zatrzymana na statku (inna niż balast), która nie jest celowo pompowana na pokład. Może gromadzić się na lub pod pokładem statku (np. pod panelami podłogowymi) w wyniku różnych mechanizmów, w tym działania fal, wycieków, poprzez gruczoły rufowe śruby napędowej oraz poprzez ładowanie przedmiotów, takich jak sprzęt do nurkowania, wędkarstwa, akwakultury lub sprzęt naukowy . W związku z tym woda zęzowa może zawierać wodę morską, jak również organizmy żywe na różnych etapach życia, szczątki komórek i zanieczyszczenia (np. za pomocą zaworów typu „kaczy dziób”. Woda zęzowa pompowana z małych statków (ręcznie lub automatycznie) zwykle nie jest oczyszczana przed odprowadzeniem do morza, w przeciwieństwie do większych statków, które są wymagane do oddzielania ropy i wody za pomocą systemów filtracji, wirowania lub absorpcji węgla. Jeśli propagule są zdolne do życia w tym procesie, zrzut wody zęzowej może spowodować rozprzestrzenienie się NIS.
W 2017 roku Fletcher i in. wykorzystali kombinację eksperymentów laboratoryjnych i terenowych do zbadania różnorodności, obfitości i przeżywalności materiału biologicznego zawartego w próbkach wody zęzowej pobranych z małych statków przybrzeżnych. Ich eksperyment laboratoryjny wykazał, że ascidian i mszywiołów larwy mogą przetrwać przejście przez niefiltrowany system pompowania w dużej mierze bez szwanku. Przeprowadzili także pierwszą ocenę morfomolekularną (przy użyciu metabarkodów eDNA) w zakresie zagrożenia bezpieczeństwa biologicznego stwarzanego przez zrzuty wody zęzowej z 30 małych jednostek pływających (żaglówek i motorówek) różnego pochodzenia i czasu żeglugi. Korzystając z metabarkodowania eDNA, scharakteryzowali około trzy razy więcej taksonów niż za pomocą tradycyjnych metod mikroskopowych, w tym wykryli pięć gatunków uznanych za nierodzime w badanym regionie.
Aby pomóc w zrozumieniu ryzyka związanego z różnymi wektorami wprowadzania NIS, tradycyjne mikroskopowe oceny bioróżnorodności są coraz częściej uzupełniane metabarkodami eDNA. Pozwala to na identyfikację szerokiej gamy różnorodnych zespołów taksonomicznych na wielu etapach życia. Może również umożliwić wykrywanie NIS, które mogły zostać przeoczone przy użyciu tradycyjnych metod. Pomimo ogromnego potencjału narzędzi metabarkodowania eDNA do badań przesiewowych taksonomicznych na szeroką skalę, kluczowym wyzwaniem dla eDNA w kontekście monitorowania środowiskowego szkodników morskich, a zwłaszcza monitorowania środowisk zamkniętych, takich jak niektóre przestrzenie zęzowe lub zbiorniki balastowe, jest różnicowanie martwych i żywych organizmy. Pozakomórkowe DNA może przetrwać w ciemnym/zimnym środowisku przez dłuższy czas (miesiące lub lata, dlatego wiele organizmów wykrytych za pomocą metabarkodowania eDNA mogło nie być zdolnych do życia w miejscu pobrania próbki przez kilka dni lub tygodni. W przeciwieństwie do tego kwas rybonukleinowy ( RNA) pogarsza się szybko po śmierci komórki, prawdopodobnie zapewniając dokładniejszą reprezentację żywotnych społeczności. Ostatnie badania metabarkodów zbadały wykorzystanie współekstrahowanych cząsteczek eDNA i eRNA do monitorowania próbek osadów bentosowych wokół morskich hodowli ryb i miejsc odwiertów ropy naftowej i wspólnie odkryli nieco silniejsze korelacje między zmiennymi biologicznymi i fizykochemicznymi wzdłuż gradientów wpływu przy użyciu eRNA.Z perspektywy bioasekuracji morskiej wykrycie żywych NIS może stanowić poważniejsze i bezpośrednie zagrożenie niż wykrycie NIS oparte wyłącznie na sygnale DNA. Środowiskowy RNA może zatem stanowić użyteczną metodę identyfikacji żywych organizmów w próbkach.
Różnorodny
Konstrukcja biblioteki genetycznych kodów kreskowych początkowo koncentrowała się na rybach i ptakach, a następnie na motylach i innych bezkręgowcach. W przypadku ptaków próbkę DNA zazwyczaj pobiera się z klatki piersiowej.
Naukowcy opracowali już specjalne katalogi dla dużych grup zwierząt, takich jak pszczoły, ptaki, ssaki czy ryby. Innym zastosowaniem jest analiza pełnej zoocenozy danego obszaru geograficznego, na przykład projekt „Polar Life Bar Code”, którego celem jest zebranie cech genetycznych wszystkich organizmów żyjących w regionach polarnych; obu biegunach Ziemi. Z tą formą związane jest kodowanie całej ichtiofauny basenu hydrograficznego, na przykład tej, która zaczęła się rozwijać w Rio São Francisco, w północno-wschodniej Brazylii .
Potencjał wykorzystania kodów kreskowych jest bardzo szeroki, od odkrycia wielu gatunków tajemniczych (przyniosło to już liczne pozytywne rezultaty), zastosowania w identyfikacji gatunków na każdym etapie ich życia, bezpiecznej identyfikacji w przypadku gatunków chronionych którymi handluje się nielegalnie itp.
Potencjały i wady

Potencjały
Zaproponowano kodowanie kreskowe DNA jako sposób na rozróżnienie gatunków, który jest odpowiedni nawet dla osób niebędących specjalistami.
niedociągnięcia
Ogólnie rzecz biorąc, wady kodów kreskowych DNA dotyczą również metabarkodów. Szczególną wadą badań metabarkodów jest to, że nie ma jeszcze zgody co do optymalnego projektu eksperymentu i kryteriów bioinformatycznych, które mają być stosowane w metabarkodowaniu eDNA. Istnieją jednak obecnie wspólne próby, takie jak sieć COST DNAqua-Net Europejskiej Współpracy w dziedzinie Nauki i Technologii , aby iść naprzód poprzez wymianę doświadczeń i wiedzy w celu ustanowienia standardów najlepszych praktyk w zakresie biomonitoringu.
Tak zwany kod kreskowy to region mitochondrialnego DNA w obrębie genu oksydazy cytochromu c . Baza danych Barcode of Life Data Systems (BOLD) zawiera sekwencje kodów kreskowych DNA z ponad 190 000 gatunków. Jednak naukowcy, tacy jak Rob DeSalle, wyrazili zaniepokojenie, że klasyczna taksonomia i kod kreskowy DNA, które uważają za mylące, muszą zostać pogodzone, ponieważ inaczej wyznaczają gatunki. Introgresja genetyczna, w której pośredniczą endosymbionty i inne wektory, może dodatkowo sprawić, że kody kreskowe będą nieskuteczne w identyfikacji gatunków.
Status gatunków kodów kreskowych
W mikrobiologii geny mogą swobodnie przemieszczać się nawet między odlegle spokrewnionymi bakteriami, prawdopodobnie obejmując całą domenę bakteryjną. Z reguły mikrobiolodzy przyjęli, że rodzaje bakterii lub archeonów z sekwencjami genów rybosomalnego RNA 16S bardziej podobnymi do siebie w ponad 97% muszą zostać sprawdzone przez hybrydyzację DNA-DNA , aby zdecydować, czy należą do tego samego gatunku, czy nie. Pojęcie to zostało zawężone w 2006 roku do podobieństwa 98,7%.
Hybrydyzacja DNA-DNA jest przestarzała, a wyniki czasami prowadziły do błędnych wniosków na temat gatunków, jak w przypadku granatowca i wydrzyka wielkiego . Nowoczesne podejścia porównują podobieństwo sekwencji za pomocą metod obliczeniowych.
Zobacz też
- Kod kreskowy systemu danych dotyczących życia (BOLD)
- Konsorcjum na rzecz kodu kreskowego życia (CBOL)
- Międzynarodowa współpraca w zakresie baz danych sekwencji nukleotydów (INSDC)
- Marker molekularny
- Przeszkoda taksonomiczna
Dalsze referencje
- ^ Santoferrara, Luciana; Burki, Fabien; Filker, Sabine; Logares, Ramiro; Dunthorn, Micheasz; McManus, George B. (2020). „Perspektywy z dziesięciu lat badań nad protistami za pomocą metabarkodowania o dużej przepustowości”. Dziennik mikrobiologii eukariotycznej . 67 (5): 612–622. doi : 10.1111/jeu.12813 . hdl : 10261/223228 . PMID 32498124 . S2CID 219331807 .