Borylowanie
Katalizowane metalem reakcje borylowania C – H to katalizowane metalem przejściowym reakcje organiczne, które wytwarzają związek borowy poprzez funkcjonalizację alifatycznych i aromatycznych wiązań C – H i dlatego są użytecznymi reakcjami do aktywacji wiązania węgiel-wodór . Katalizowane metalem reakcje borylowania C – H wykorzystują metale przejściowe do bezpośredniego przekształcenia wiązania C – H w wiązanie C – B. Ta droga może być korzystna w porównaniu z tradycyjnymi reakcjami borylowania, wykorzystując tani i występujący w dużych ilościach węglowodorowy materiał wyjściowy, ograniczając prefunkcjonalizowane związki organiczne, redukując toksyczne produkty uboczne i usprawniając syntezę biologicznie ważnych cząsteczek. Kwasy boronowe i estry boronowe to powszechne grupy borylowe włączane do cząsteczek organicznych w reakcjach borylowania. Kwasy boronowe to trójwartościowe związki organiczne zawierające bor, które posiadają jeden podstawnik alkilowy i dwie grupy hydroksylowe. Podobnie estry boronowe mają jeden podstawnik alkilowy i dwie grupy estrowe. Kwasy i estry boronowe są klasyfikowane w zależności od rodzaju grupy węglowej (R) bezpośrednio związanej z borem, na przykład estry alkilowe, alkenylowe, alkinylowe i aryloborowe. Najbardziej powszechny rodzaj materiałów wyjściowych, które włączają estry boronowe do związków organicznych w reakcjach borowania katalizowanych przez metale przejściowe, mają wzór ogólny (RO) 2B -B(OR) 2 . Na przykład bis(pinakolato)dibor (B 2 Pin 2 ) i bis(katecholato)diboran (B 2 Cat 2 ) są powszechnymi źródłami boru o tym ogólnym wzorze.

Atom boru w estrze lub kwasie boronowym jest zhybrydyzowany sp2 z wolnym orbitalem p, co umożliwia tym grupom działanie jak kwasy Lewisa . Wiązania C-B kwasów boronowych i estrów są nieco dłuższe niż typowe wiązania pojedyncze C-C w zakresie 1,55-1,59 Å. Wydłużone wiązanie C-B w stosunku do wiązania C-C powoduje, że energia wiązania jest również nieco mniejsza niż energia wiązań C-C (323 kJ/mol dla C-B w porównaniu z 358 kJ/mol dla C-C). Wiązanie węgiel-wodór ma długość wiązania około 1,09 Å i energię wiązania około 413 kJ/mol. Wiązanie C – B jest zatem użytecznym związkiem pośrednim jako wiązanie, które zastępuje typowo niereaktywne wiązanie C – H.
borowoorganiczne to związki organiczne zawierające wiązanie węgiel-bor. Związki boroorganiczne mają szerokie zastosowanie w syntezie chemicznej, ponieważ wiązanie C – B można łatwo przekształcić w wiązanie C – X ( X = Br, Cl), C – O, C – N lub C – C. Ze względu na wszechstronność wiązań C – B opracowano wiele procesów włączania ich do związków organicznych. Związki boroorganiczne są tradycyjnie syntetyzowane z odczynników Grignarda poprzez reakcje hydroborowania lub diborowania. Borylacja stanowi alternatywę.
Katalizowane metalem reakcje borylowania C – H
Alifatyczne borylowanie C – H
Jak po raz pierwszy opisał Hartwig, alkany mogą być selektywnie borylowane z wysoką selektywnością dla pierwszorzędowego wiązania C–H przy użyciu Cp*Rh(η4 - C6Me6 ) jako katalizatora . Warto zauważyć, że selektywność dla pierwszorzędowego wiązania C – H jest wyłączna nawet w obecności heteroatomów w łańcuchu węgiel-wodór. Katalizowane rodem borylowanie wiązań metylowych C-H zachodzi selektywnie, bez zależności od położenia heteroatomu. Borylacja zachodzi selektywnie przy najmniejszym zakłóceniu przestrzennym i najmniej bogatym w elektrony pierwotnym wiązaniu C – H w szeregu acetali , eterów , amin i fluorków alkilu. Ponadto wykazano, że żadna reakcja nie zachodzi przy braku pierwszorzędowych wiązań C – H, na przykład gdy substratem jest cykloheksan .

Selektywna funkcjonalizacja pierwszorzędowego wiązania alkanu jest spowodowana tworzeniem korzystnego kinetycznie i termodynamicznie pierwszorzędowego kompleksu alkil-metal w stosunku do tworzenia drugorzędowego kompleksu alkil-metal.

Większą stabilność pierwszorzędowych i drugorzędowych kompleksów alkilowych można przypisać kilku czynnikom. Po pierwsze, pierwszorzędowy kompleks alkilowy jest uprzywilejowany sterycznie nad drugorzędowym kompleksem alkilowym. Po drugie, częściowe ładunki ujemne są często obecne na węglu α kompleksu metal-alkil, a pierwszorzędowy ligand alkilowy podtrzymuje częściowy ładunek ujemny lepiej niż drugorzędowy ligand alkilowy. Pochodzenie selektywności alifatycznego borylowania C – H przy użyciu katalizatorów rodowych badano za pomocą rodzaju badania mechanistycznego zwanego wymianą wodór-deuter . Wymiana H / D wykazała, że regioselektywność całego procesu pokazanego poniżej wynika z selektywnego rozszczepienia pierwszorzędowych wiązań C – H i selektywnej funkcjonalizacji pierwszorzędowego związku pośredniego metal-alkil w stosunku do drugorzędowego związku pośredniego metal-alkil.

Syntetyczna użyteczność alifatycznego borylowania C – H została zastosowana do modyfikacji polimerów poprzez borylowanie, a następnie utlenianie w celu utworzenia polimerów z grupami hydroksylowymi.

Aromatyczne borylowanie C – H
Steryczne borylowanie C – H arenów
Pierwszy przykład katalitycznego borylowania C – H nieaktywowanego węglowodoru (benzenu) opisali Smith i Iverson przy użyciu Ir (Cp *) (H) (Bpin) jako katalizatora. Wydajność tego układu była jednak niska, zapewniając jedynie 3 obroty po 120 hw temperaturze 150°C. Liczne późniejsze osiągnięcia Hartwiga i współpracowników doprowadziły do skutecznych, praktycznych warunków borylowania areny. Aromatyczna borylacja C-H została opracowana przez Johna F. Hartwiga i Ishiyamę przy użyciu odczynnika diborowego Bis(pinacolato)diboronu katalizowanego przez 4,4'-di-tert-butylobipirydynę (dtbpy) i [Ir(COD)(OMe)] 2 . W przypadku tego układu katalizatorów borylowanie aromatycznych wiązań C-H zachodzi z regioselektywnością, która jest kontrolowana przez efekty steryczne początkowego arenu. Selektywność funkcjonalizacji aromatycznych wiązań C-H jest regulowana ogólną zasadą, że reakcja nie zachodzi w pozycji orto do podstawnika, gdy dostępne jest wiązanie C-H pozbawione podstawnika w pozycji orto . Gdy obecna jest tylko jedna grupa funkcyjna, borylacja zachodzi w pozycji meta i para w stosunku statystycznym 2:1 (meta:para). Orto względu na efekty steryczne podstawnika.

Dodanie Bpin występuje tylko w jednej pozycji dla symetrycznie podstawionych 1,2- i 1,4-podstawionych arenów. Symetryczne lub niesymetryczne 1,3-podstawione areny są również selektywnie borylowane, ponieważ tylko jedno wiązanie C – H jest dostępne sterycznie.

Kontrastuje to z elektrofilową substytucją aromatyczną , w której regioselektywnością rządzą efekty elektroniczne.

Syntetyczne znaczenie aromatycznego borylowania C – H pokazano poniżej, gdzie 1,3-dipodstawiony związek aromatyczny można bezpośrednio przekształcić w związek 1,3,5-organoboranu, a następnie funkcjonalizować.

Aromatyczna funkcjonalizacja C – H została z powodzeniem włączona do całkowitej syntezy Complanadyny A, alkaloidu Lycopodium , który zwiększa ekspresję mRNA dla czynnika wzrostu nerwów (NGF) i produkcję NGF w ludzkich komórkach glejowych . Naturalne produkty, które promują rozwój nowych sieci neuronowych, są interesujące w leczeniu chorób takich jak choroba Alzheimera . Komplanadyna A została pomyślnie zsyntetyzowana przy użyciu kombinacji bezpośredniego aromatycznego borylowania C – H opracowanego przez Hartwiga i Ishyiamę, a następnie sprzęgania krzyżowego Suzuki – Miyaura , a następnie rozszczepienia grupy zabezpieczającej Boc .

Borylowanie C – H heteroarenów
Heteroareny mogą również ulegać borylacji w warunkach katalizowanych irydem, jednak selektywność miejscowa w tym przypadku jest kontrolowana przez efekty elektroniczne , w których furany , pirole i tiofeny ulegają reakcji przy wiązaniu C – H alfa z heteroatomem. W tym przypadku sugeruje się, że selektywność występuje poprzez wiązanie C – H alfa do heteroatomu, ponieważ jest to najbardziej kwaśne wiązanie C – H, a zatem najbardziej reaktywne.

Ukierunkowana borylacja orto C – H
Stosując ten sam układ katalizatorów grupy kierujące można zastosować w celu uzyskania regioselektywności bez podstawników jako mediatorów sterycznych. Na przykład Boebel i Hartwig opisali metodę przeprowadzania orto -borylacji, w której grupa kierująca dimetylohydrosililem na arenie ulega katalizowanej irydem borylacji przy wiązaniu C – H orto do grupy kierującej silanem . Selektywność dla orto w przypadku stosowania hydrosililowych grup kierujących została przypisana odwracalnemu dodaniu wiązania Si-H do centrum metalu, co prowadzi do preferencyjnego rozszczepienia wiązania C-H orto do podstawnika hydrosililowego. Kilka innych strategii osiągnięcia orto -borylacji arenów zostało opracowanych przy użyciu różnych grup kierujących.

Mechanistyczny szczegół borlyacji arenów C – H
Zaproponowano kompleks trisboryloirydu w celu ułatwienia mechanizmu każdej z tych reakcji, które skutkują borylacją C – H arenów i heteroarenów. Badania kinetyczne i badania znakowania izotopowego wykazały, że kompleks triborylowy Ir(III) reaguje z arenem w procesie katalitycznym. Poniżej przedstawiono wersję cyklu katalitycznego dla orto- borylowania związków hydrosilanowych. Dane kinetyczne pokazują, że obserwowany kompleks trisborylowy skoordynowany z cyklooktenem szybko i odwracalnie dysocjuje cyklookten, tworząc 16-elektronowy kompleks trisborylowy. W przypadku zastosowania benzylodimetylosilanu jako grupy kierującej proponuje się, aby benzylodimetylosilan reagował z katalizatorem trisboryloirydowym poprzez odwracalną addycję wiązania Si-H do centrum metalu, a następnie selektywną aktywację wiązania orto-C-H poprzez addycję utleniającą i redukcyjną eliminacja .

Meta-selektywna borylacja : Meta-selektywna borylacja C – H to ważna transformacja syntetyczna, którą odkrył w 2002 roku Smith III z Michigan State University w USA. Jednak to metaborylowanie było całkowicie sterycznie sterowane i ograniczało się tylko do 1,3-dipodstawionych benzenów. Około 12 lat później dr Chattopadhyay i jego zespół z Centrum Badań Biomedycznych w UP w Indiach odkryli elegancką technologię metaselektywnej aktywacji i borylacji wiązań C–H. Zespół wykazał, że przy użyciu tego samego substratu można zmienić selektywność pozycyjną na drugą, po prostu zmieniając ligand. Pochodzenie metaselektywności zostało określone przez dwa parametry, takie jak: 1) oddziaływanie elektrostatyczne, 2) wtórne oddziaływanie BN.
W tym samym czasie zespół z Japonii, dr Kanai, przedstawił niesamowitą koncepcję metaselektywnej borylacji opartej na interakcji wtórnej. Metoda ta obejmuje borowanie różnych związków karbonylowych.
Reakcje redukcji ze związkami borowoorganicznymi
Redukcja Corey – Bakshi – Shibata (redukcja CBS)
W 1981 roku Hirao i współpracownicy odkryli, że asymetryczna redukcja prochiralnych ketonów aromatycznych za pomocą chiralnych aminoalkoholi i boranu dała odpowiednie alkohole drugorzędowe o 60% ee . Odkryli, że chiralne aminoalkohole reagują z boranem , tworząc kompleksy aloksylo-amina-boran. Proponuje się, aby kompleksy zawierały stosunkowo sztywny pięcioczłonowy układ pierścieni, co czyni je stabilnymi termicznie i hydrolitycznie oraz rozpuszczalnymi w szerokiej gamie protonowych i aprotonowych.
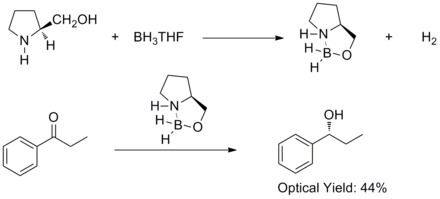
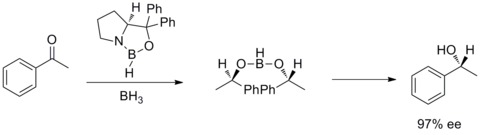
W 1987 roku Elias James Corey i współpracownicy odkryli, że tworzenie się oksazaborolidyn z boranu i chiralnych aminoalkoholi. Stwierdzono, że oksazaborolidyny katalizują szybką i wysoce enancjoselektywną redukcję prochiralnych ketonów w obecności BH3THF. Ta enancjoselektywna redukcja ketonów achiralnych za pomocą katalitycznej oksazaborolidyny nazywana jest redukcją Corey – Bakshi – Shibata lub redukcją CBS.
Redukcja Midland Alpine-boran (redukcja Midland)
W 1977 roku MM Midland i współpracownicy donieśli o zaskakującej obserwacji, że B-3-alfa-pinanylo-9-borabicyklo[3,3,1]nonan, łatwo otrzymany przez hydroborowanie (+)-alfa-pinenu z 9- borobicyklo [3,3,1]nonan szybko redukuje benzaldehyd-alfa-d do alkoholu (S)-(+)-benzylo-alfa-d z zasadniczo ilościową asymetryczną indukcją.
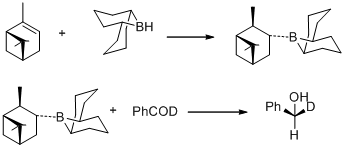
W tym samym roku MM Midland odkrył B-3-alfa-pinanylo-9-BBN jako środek redukujący, który może być łatwo dostępny w reakcji (+)-alfa-pinenu z 9-BBN. Nowy środek redukujący został później skomercjalizowany przez Aldrich Co. pod nazwą Alpine Borane , a asymetryczna redukcja grup karbonylowych za pomocą dowolnego enancjomeru Alpine-Boran jest znana jako redukcja Midland Alpine-Boran.
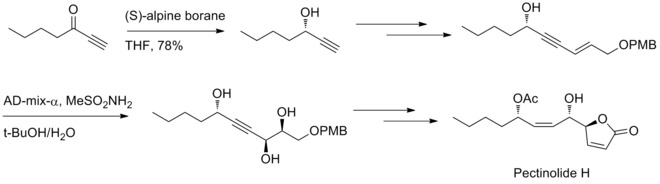
W 2012 roku URY Venkateswarlu i współpracownicy opisali stereoselektywną metodę syntezy pektynolidu H. Redukcja Midlanda i reakcja dihydroksylacji Sharplessa są zaangażowane w generowanie trzech centrów chiralnych na C-4 ', C-5 i C-1'.
Reakcje sprzęgania ze związkami borowoorganicznymi
Reakcja Mannicha z kwasem Petasis boronowym
W 1993 roku NA Petasis i I. Akrltopoulou opisali wydajną syntezę amin allilowych w zmodyfikowanej reakcji Mannicha . W tej zmodyfikowanej reakcji Mannicha odkryli, że kwasy winyloborowe mogą uczestniczyć jako nukleofile , dając geometrycznie czyste alliloaminy. Ta zmodyfikowana reakcja Mannicha była znana jako reakcja Mannicha z kwasem boronowym Petasisa.
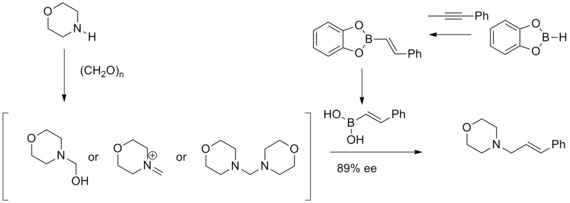
Asymetryczna alicja Rousha
W 1978 roku RW Hoffmann i T. Herold opisali enancjoselektywną syntezę drugorzędowych alkoholi homoallilowych poprzez chiralne nieracemiczne estry alliloboronowe . Alkohole homoallilowe powstały z doskonałą wydajnością i umiarkowaną enancjoselektywnością.
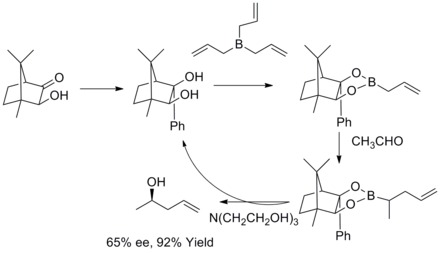
W 1985 roku WR Roush i współpracownicy odkryli, że boraniany allilowe modyfikowane winianami oferują proste, bardzo atrakcyjne podejście do kontroli selektywności twarzy w reakcjach z chiralnymi i achiralnymi aldehydami. W kolejnych latach WR Roush i współpracownicy rozszerzyli tę strategię na syntezę but-2-eno-1,4-dioli i antydioli. Ten rodzaj reakcji jest znany jako asymetryczne allilowanie Roucha.
W 2011 roku RA Fernandes i P. Kattanguru ukończyli ulepszoną całkowitą syntezę diastereoizomerów (8S, 11R, 12R) - i (8R, 11R, 12R) -topsentolidu B2 w ośmiu etapach. W pracy wykorzystano diastereoselektywną reakcję allilowania Rousha jako kluczową reakcję w syntezie całkowitej polegającej na wprowadzeniu dwóch chiralnych związków pośrednich. Następnie autorzy zsyntetyzowali dwa diastereoizomery poprzez te dwa chiralne związki pośrednie.

Sprzęg krzyżowy Suzuki – Miyaura
W 1979 roku N. Miyaura i A. Suzuki opisali syntezę arylowanych (E)-alkenów z wysoką wydajnością z halogenków arylowych z alkilo-1-enyloboranami i katalizowaną tetrakis( trifenylofosfino )palladem i zasadami. Następnie A. Suzuki i współpracownicy rozszerzyli ten rodzaj reakcji na inne związki borowe i inne halogenki alkenylu, arylu , alkilu i triflat . Reakcja sprzęgania krzyżowego katalizowana palladem związki borowoorganiczne i te halogenki organiczne tworzące wiązania węgiel-węgiel są znane jako sprzęganie krzyżowe Suzuki – Miyaura .
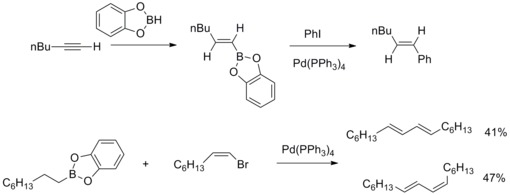
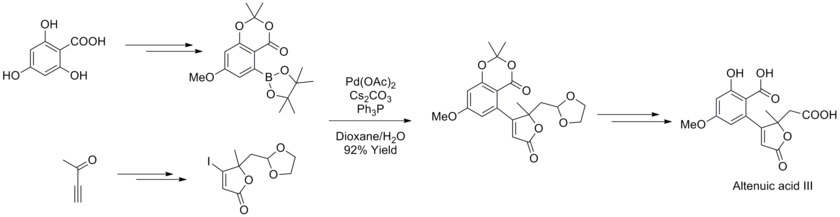
W 2013 roku Joachim Podlech i współpracownicy określili strukturę Alternaria mycotoxin altenuic acid III za pomocą analizy spektroskopowej NMR i zakończyli jego całkowitą syntezę. W strategii syntezy zastosowano reakcję sprzęgania krzyżowego Suzuki-Miyaura z wysoce funkcjonalizowanym boronianem i butenolidami w celu syntezy prekursora produktu naturalnego z wysoką wydajnością.
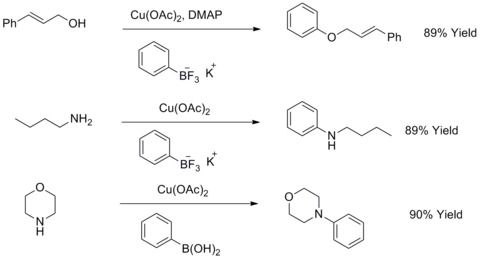
Synteza modyfikowanego eteru biarylowego Ullmanna i aminy biarylowej
W 1904 roku Fritz Ullmann odkrył, że proszek miedzi może znacznie poprawić reakcję halogenków arylowych z fenolami, dając etery biarylowe. Ta reakcja jest znana jako kondensacja Ullmanna . W 1906 roku I. Goldberg rozszerzył tę reakcję, aby zsyntetyzować aryloaminę w reakcji halogenków arylu z amidem w obecności węglanu potasu i CuI. Ta reakcja jest znana jako kondensacja Ullmanna zmodyfikowana przez Goldberga. W 2003 roku RA Batey i TD Quach zmodyfikowali ten rodzaj reakcji, stosując sole trifluoroboranów potasu do reakcji z alkoholami alifatycznymi, aminami alifatycznymi lub anilinami w celu syntezy eterów arylowych lub amin arylowych.
Zobacz też
- Chemia borowoorganiczna
- Reakcje organoboranów i boranów
- Redukcja Coreya-Itsuno
- Redukcja boranu z regionu Midland Alpine
- Reakcja petasisa
- Reakcja Suzukiego