choroba Olliera
 | |
| Zdjęcie rentgenowskie kości promieniowej 7-letniej dziewczynki z chorobą Olliera. | |
| choroby Olliera przedstawiające enchondromy zlokalizowane w dolnej części | |
|---|---|
| Specjalność |
Genetyka medyczna |
Choroba Olliera jest rzadką, sporadyczną, niedziedziczną chorobą szkieletu, w której zwykle w pobliżu chrząstki płytki wzrostowej rozwijają się łagodne guzy chrzęstne ( enchondromy ). Jest to spowodowane resztkami chrząstki, które rosną i znajdują się w przynasadzie lub trzonie i ostatecznie mineralizują z czasem, tworząc liczne enchondromy. Do głównych objawów tego zaburzenia należy asymetria i skrócenie kończyny oraz zwiększona grubość brzegu kostnego. Objawy te są zwykle widoczne po raz pierwszy we wczesnym dzieciństwie, a średni wiek rozpoznania wynosi 13 lat. Wielu pacjentów z chorobą Olliera jest podatnych na rozwój innych nowotworów złośliwych, w tym kości mięsaki wymagające leczenia i usunięcia złośliwego nowotworu kości. Przypadki u pacjentów z chorobą Olliera wykazały związek z mutacjami genów IDH1, IDH2 i PTH1R. Obecnie nie ma form leczenia choroby Olliera, ale powikłania, takie jak złamania, deformacje, nowotwory, które z niej wynikają, można leczyć za pomocą zabiegów chirurgicznych. Częstość występowania tego schorzenia szacuje się na około 1 przypadek na 100 000. Nie jest jasne, czy mężczyźni czy kobiety są bardziej dotknięci tym zaburzeniem ze względu na sprzeczne studia przypadków.
Prezentacja
Choroba polega na narastaniu mnogich enchondroma , które zwykle rozwijają się we wczesnym dzieciństwie. Wzrost tych enchondroma zwykle zatrzymuje się po dojrzewaniu szkieletu. Dotknięta kończyna jest skrócona (karłowatość asymetryczna) i czasami wygięta z powodu anomalii fuzji nasad. Zmiany kostne na ogół w dzieciństwie mają charakter komórkowy i z czasem stają się bardziej pojedyncze. Osoby z chorobą Olliera są podatne na łamanie kości ( złamania ) i zwykle mają opuchnięte, obolałe kończyny. Jednak wiele przypadków samotnych enchondromatów pozostaje niezauważonych z powodu braku objawów. Enchondromy są powszechnie spotykane w paliczkach, kościach śródręcza i śródstopia u pacjentów z chorobą Olliera ze względu na powinowactwo enchondroma do długich rurkowatych kości, takich jak kość udowa i kość ramienna. Zwykle obserwuje się jednostronne rozmieszczenie zmian kostnych, ale mogą również wystąpić obustronne rozmieszczenie zmian lub pojedyncza kończyna. Około jedna trzecia przypadków wykazuje jakąś formę fizycznych deformacji polegających na wygięciu lub nieprawidłowym wydłużeniu kończyn. [ potrzebne źródło ]
Powiązane warunki
Choroba Olliera wiąże się z większym ryzykiem nowotworów złośliwych, takich jak ośrodkowy układ nerwowy (OUN), jajnik i gruczolakorak . Glejaki czaszki zostały powiązane z tym zaburzeniem ze zwiększoną częstością i we wcześniejszym wieku rozpoznania. Większość przypadków glejaka zawiera mutacje genu IDH, co wyjaśnia związek między tymi dwoma stanami. Z chorobą wiąże się również nowotwór z młodzieńczych komórek warstwy ziarnistej. Jedno studium przypadku wskazuje, że jest to spowodowane dysplazją mezodermalną w kościach długich, prowadzącą do raka jajnika.
Częstość występowania wtórnego chrzęstniakomięsaka w chorobie Olliera jest najczęściej szacowana na 25–30%, a niektóre prognozy sięgają nawet 50%. Chrzęstniakomięsaki zwykle rozwijają się w młodym wieku dorosłym i najczęściej tworzą się jako jednoogniskowa dystrybucja u pacjentów z chorobą Olliera. Najczęstsze lokalizacje guzów to miednica i obręcz barkowa. Chociaż chrzęstniakomięsak jest najczęstszą postacią wtórnego nowotworu złośliwego kości występującego w przypadku choroby Olliera, mogą wystąpić inne formy, takie jak struniaki i kostniakomięsaki. Nieleczone te złośliwe przemiany mogą prowadzić do zgonu. [ potrzebne źródło ]
Pokrewne, a nawet rzadsze zaburzenie zwane zespołem Maffucciego jest bardzo podobnym stanem, który charakteryzuje się obecnością wielu enchondroma z naczyniakami krwionośnymi i czasami naczyniakami limfatycznymi, zwykle w pobliżu dłoni i stóp, ale nie ogranicza się do czaszki, żeber i kości kręgosłupa. To zaburzenie jest również sporadyczne i niedziedziczne i zwykle wykrywane w dzieciństwie, mimo że jest stanem wrodzonym. Zespół Maffucciego został również podobnie powiązany z mutacjami IDH1 i IDH2, w szczególności z mutacją hotspot IDH1 R132C. Przypuszcza się, że ta mutacja hotspot jest odpowiedzialna za naczyniaki krwionośne i enchondromy komórek wrzecionowatych w przypadkach zespołu Maffucciego. Analiza sekwencjonowania Sangera wykazała, że ekson 4 jest główną lokalizacją mutacji w genach IDH1 i IDH2, które są szczególnie odpowiedzialne za naczyniaki krwionośne. Zespół Maffucciego niesie ze sobą znacznie większe ryzyko przemian złośliwych, takich jak chrzęstniakomięsaki, ale także znacznie bardziej agresywne nowotwory, takie jak ostra białaczka limfocytowa oraz nowotwory przewodu pokarmowego i jajnika.
Przyczyna
Przez wiele lat większość badań była niejednoznaczna co do przyczyny choroby.
Ostatnie badania wykazały, że uważa się, że większość przypadków choroby Olliera była spowodowana mutacjami dehydrogenaz izocytrynianowych IDH1 i IDH2. W jednym badaniu 35 z 43 (81%) pacjentów z chorobą Olliera miało mutację IDH1 lub IDH2. Inne badanie sugeruje, że mutacje R132C IDH1, które są szczególnie dominujące w eksonie 4 genów IDH, są powiązane ze wzrostem zmian naczyniowych. Deyhydrogenazy izocytrynianowe IDH1 i IDH2 są katalizatorami odpowiedzialnymi za konwersję izocytrynianu do 2-oksoglutaranu. Mutacje deyhydrogenaz izocytrynianowych IDH1 i IDH2 zakłócają ten proces, powodując nieuregulowaną produkcję α-ketoglutaranu i zmniejszenie proliferacji chondrocytów. W wielu przypadkach guzów chrzęstnych stwierdzono mutacje punktowe IDH1 i IDH2, co wyjaśnia, dlaczego choroba Olliera jest związana z wieloma różnymi powiązanymi stanami. Opierając się na tych studiach przypadków, większość dowodów sugeruje, że nieprawidłowa wyściółka zmian stwierdzona w chorobie Olliera sugerowałaby, że stan ten jest spowodowany postzygotyczną mutacją somatyczną, co prowadzi do mozaikowego zaburzenia genetycznego.
Około 8–10% przypadków pacjentów z chorobą Olliera zostało powiązanych z mutacjami PTH1R. Szczególne studium przypadku zmutowanego heterozygotycznego receptora PTHR1 (R150C) zaobserwowano u dwóch niespokrewnionych pacjentów z chorobą Olliera. Ten mutant PTHR1 (R150C) powoduje zmniejszenie różnicowania chondrocytów poprzez uruchomienie szlaku zależnego od PTHrP i zmniejszenie funkcji receptora PTHLH o około 30%, tworząc enchondromy. Stwierdzono, że jeden z tych pacjentów z mutantem PTHR1 (R150C) odziedziczył mutację po swoim ojcu. Daje to wiarę teorii, że do wystąpienia choroby Olliera potrzeba wielu mutacji genetycznych.
Alternatywna teoria sugeruje, że skoro zdarzały się przypadki enchondromatozy u wielu członków rodziny, zaburzenie to może być przekazywane w drodze dziedziczenia autosomalnego dominującego.
Diagnoza
Ocenę kliniczną i radiologiczną przeprowadza się w celu wykrycia obecności nowotworów kości lub zmian typowych dla choroby Olliera. Oceny histologiczne są wykorzystywane głównie do badania lub wykrywania nowotworów złośliwych.
U pacjentów z chorobą Olliera można zaobserwować nieprawidłowy wzrost kości, taki jak skrócenie lub pogrubienie i deformację. Te zmiany kostne są widoczne przy urodzeniu za pomocą radiografii, ale zwykle nie są poddawane badaniom przesiewowym ani badanym, dopóki nie pojawią się objawy kliniczne we wczesnym dzieciństwie. Jednak niektórzy pacjenci mogą nie wykazywać żadnych objawów. W jednym badaniu stwierdzono, że średni wiek rozpoznania u pacjentów z chorobą Olliera to trzynaście lat. Na zdjęciu rentgenowskim zwykle występuje kilka jednorodnych zmian o owalnym lub wydłużonym kształcie z nieznacznie pogrubionymi krawędziami kości. Z wiekiem zmiany te mogą ulegać zwapnieniu i wyglądać jak rozproszone, drobne plamki lub cętki. Wachlarzowe przegrody lub smugi wskazywałyby na obecność kilku enchondromów. Wczesne wykrywanie oraz konsekwentne i powtarzane monitorowanie jest ważne dla zapobiegania i leczenia wszelkich potencjalnych nowotworów kości. [ potrzebne źródło ]
Rezonans magnetyczny (MRI), ultrasonografia i scyntygrafia na ogół nie są praktyczne do celów diagnostycznych. Promienie rentgenowskie nie są tak skuteczne w monitorowaniu lub ocenie enchondromów ze względu na częste zlokalizowane zmiany, czasami również z powodu dużej liczby enchondromów. Czasami jednak MRI może być wykorzystany do monitorowania i oceny zmian objawowych w przypadku potencjalnych przemian złośliwych.
Podobne zaburzenia, takie jak zespół Maffucciego i dziedziczne mnogie egzostozy (HME), wymagają różnicowania podczas diagnozy. Zespół Maffucciego można odróżnić klinicznie na podstawie obecności naczyniaków krwionośnych i naczyniaków limfatycznych oraz genetycznie na podstawie mutacji hotspot R132C IDH1. W HME występują kostniakochrzęstniaki, które znajdują się blisko powierzchni kości, podczas gdy enchondroma występujące w chorobie Olliera i Maffucciego są bardziej w kierunku środka kości. Również uciski nerwowe są częściej spotykane w HME niż w chorobie Olliera.
Leczenie
Stanu choroby Olliera nie można leczyć, ale pojawiające się powikłania, takie jak złamania, wady wzrostu i guzy, można leczyć chirurgicznie. Są one zwykle wykonywane w celu leczenia i usunięcia wszelkich obcych tkanek kostnych przy jednoczesnym zachowaniu funkcji kończyny, jeśli to możliwe.
Złamania leczono różnymi metodami, takimi jak przeszczepy kostne, stabilizacja wewnętrzna , kortykoplastyka, technika Ilizarowa, system elastycznych stabilnych gwoździ śródszpikowych (ESIN) i elastyczne gwoździe śródszpikowe (FIN).
Wykazano, że kortykoplastyka odnosi sukcesy w leczeniu uszkodzeń i deformacji rąk przy jednoczesnym zachowaniu normalnej funkcji. Operacja polega na usunięciu tkanki ( kiretaż ) i odbudowie kości w celu usunięcia enchondroma i poprawy wyglądu kosmetycznego. W wielu przypadkach kortykoplastyki wykazano poprawę wyglądu przy zachowaniu funkcji. W niektórych przypadkach obserwowano nawroty enchondroma. W przypadku choroby Olliera wskazane jest wczesne leczenie chirurgiczne enchondroma ręki.
Technika Ilizarowa jest formą nieinwazyjnego leczenia, która czasami może być stosowana do przekształcania i korygowania deformacji i niewspółosiowości kości kończyn. Wykorzystuje proces zewnętrznego mocowania poprzez rusztowanie szpilek lub drutów w kości, próbując przekształcić enchondromata w normalną tkankę kostną. Ta metoda leczenia jest bardzo bezpieczna, ale także bardzo uciążliwa, ponieważ jest to procedura długoterminowa. Wykazano, że stabilizatory pierścieniowe stosowane w technice Ilizarowa są bardzo wszechstronne w ustawianiu i wydłużaniu kości kończyn, a także w zarządzaniu napięciem tkanek miękkich. Powikłania tej procedury mogą obejmować nawrót powikłań związanych z wydłużaniem kończyn, takich jak przedwczesne gojenie wymagające osteoklasty i powrót deformacji kątowych. Aby zapobiec przedwczesnemu gojeniu, zachęca się do skrócenia okresów latencji, po których następują krótsze czasy rozproszenia uwagi. Wykazano, że technika Ilizarowa ma pozytywne wyniki w wielu przypadkach choroby Olliera, a niektórzy pacjenci doświadczają nawet pełnej korekcji deformacji i długości w najlepszych scenariuszach.
System elastycznych stabilnych gwoździ śródszpikowych (ESIN) i elastyczne gwoździe śródszpikowe (FIN) to nowsze procedury chirurgiczne wykorzystujące fiksację wewnętrzną, która, jak wykazano, zmniejsza wskaźnik gojenia i minimalizuje powikłania. Podobnie jak technika Ilizarowa zabieg ma na celu skorygowanie deformacji i wydłużenie kości kończyn. Ta forma leczenia nie nadaje się do wydłużania kości o małej średnicy trzonu lub odcinków zawierających otwarte strefy wzrostu. Pacjenci z ciężkimi postaciami choroby Olliera również nie kwalifikują się do operacji ze względu na zwiększone ryzyko powikłań z powodu niestabilności szkieletu kostnego. Zarówno system elastycznego stabilnego gwoździa śródszpikowego (ESIN), jak i elastycznego gwoździa śródszpikowego (FIN) wykorzystują dwa wygięte gwoździe elastyczne, aby umożliwić większe wyrównanie i stabilność. Wykazano, że ta technika stosowana w połączeniu z okrągłym stabilizatorem zewnętrznym znacznie zmniejsza wartości wskaźnika gojenia zarówno w wydłużaniu jednosegmentowym, jak i wielosegmentowym. Kolejną zaletą jest to, że nie stwierdzono złamań ani deformacji w późniejszych kontrolach trwających od 2 do 5 lat.
Elastyczny stabilny system gwoździ śródszpikowych (ESIN) wymaga dłuższego czasu mocowania zewnętrznego w przypadku większości zabiegów w porównaniu z elastycznym gwoździem śródszpikowym (FIN). [ potrzebne źródło ]
Złośliwe transformacje jednego lub wielu enchondromów są powszechne u pacjentów z chorobą Olliera i zwykle pojawiają się u młodych dorosłych, często wymagających operacji . Średni wiek pacjentów z chorobą Olliera do pierwszej operacji leczenia ich chrzęstniakomięsaka wynosi trzydzieści trzy lata. Niektóre przykłady zabiegów chirurgicznych wykonywanych w celu leczenia wtórnych złośliwych nowotworów kości w przebiegu choroby Olliera obejmują amputację, rozległe wycięcia, hemipelwektomię i artroplastykę. Kobalt i chemioterapia zazwyczaj nie są podstawowymi metodami chirurgicznymi, ponieważ chrzęstniakomięsaki na ogół nie mają wystarczającego dopływu krwi, aby była to skuteczna forma leczenia. Są one wykonywane w celu leczenia i usunięcia wszelkich obcych tkanek kostnych przy jednoczesnym zachowaniu funkcji kończyny, jeśli to możliwe.
Epidemiologia
Szacuje się, że jedna na 100 000 osób cierpi na chorobę Olliera. Jednak szacunki te mogą być niskie ze względu na zbyt małą liczbę zgłaszanych przypadków bezobjawowych lub łagodnych stanów. Istnieje wiele sprzecznych badań, które podają różne wskaźniki rozpowszechnienia między płciami, ponieważ jedno badanie wskazuje, że dotyka obie płci w równym stopniu, podczas gdy inne badania twierdzą, że częściej dotyka mężczyzn lub kobiety. Średni wiek rozpoznania wynosi 13 lat. Choroba Olliera jest zwykle diagnozowana dopiero we wczesnym dzieciństwie ze względu na brak widocznych objawów obecnych przy urodzeniu, pomimo obecności zmian w badaniu radiologicznym. Chociaż większość badań sugeruje, że choroba Olliera jest spontaniczna i niedziedziczna, istnieją przypadki, w których pojawia się wśród członków rodziny.
eponimy
Zaburzenie zostało nazwane na cześć francuskiego chirurga Louisa Léopolda Olliera . Pod koniec XIX wieku Ollier był jednym z pierwszych, którzy odróżnili enchondromatozę od tego stanu, podkreślając wzór nieprawidłowego i asymetrycznego rozmieszczenia enchondroma w chorobie Olliera.
Galeria
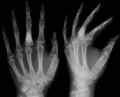
Zdjęcie rentgenowskie przedstawiające zwapniałe enchondromy zlokalizowane w palcu 37-letniego pacjenta dotkniętego chorobą Olliera

Zdjęcie rentgenowskie ukazujące enchondromy zlokalizowane w kości ramiennej 37-letniego pacjenta dotkniętego chorobą Olliera

Zdjęcie rentgenowskie ukazujące enchondromy zlokalizowane w dolnej części kości promieniowej 37-letniego pacjenta dotkniętego chorobą Olliera
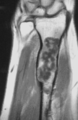
Rezonans magnetyczny ukazujący enchondromy zlokalizowane w dolnej części kości promieniowej u 37-letniej pacjentki z chorobą Olliera

Rezonans magnetyczny ukazujący enchondromy zlokalizowane w dolnej części kości promieniowej u 37-letniego pacjenta z chorobą Olliera.

Enchondroma zlokalizowane w górnej części kości ramiennej u tej samej pacjentki





