inhibitor c-Met
Inhibitory c-Met to klasa małych cząsteczek , które hamują aktywność enzymatyczną kinazy tyrozynowej c-Met , receptora czynnika wzrostu hepatocytów/czynnika rozproszenia (HGF/SF). Inhibitory te mogą mieć zastosowanie terapeutyczne w leczeniu różnych typów nowotworów.
Wiele inhibitorów c-Met jest obecnie [ kiedy? ] w badaniach klinicznych . Kryzotynib i kabozantynib jako pierwsze zostały zatwierdzone przez amerykańską FDA . Kryzotynib uzyskał przyspieszoną rejestrację w 2011 roku do leczenia pacjentów z miejscowo zaawansowanym lub przerzutowym niedrobnokomórkowym rakiem płuca , podczas gdy kabozantynib został zatwierdzony w 2012 roku do leczenia rdzeniastego raka tarczycy i rozpoczął również badania kliniczne w leczeniu kilku innych typów raka.
c-Met stymuluje rozpraszanie komórek, inwazję, ochronę przed apoptozą i angiogenezą . c-Met jest receptorową kinazą tyrozynową , która może powodować wiele różnych nowotworów, takich jak rak nerki , żołądka i drobnokomórkowy rak płuc , nowotwory ośrodkowego układu nerwowego , a także kilka mięsaków , gdy jej aktywność jest rozregulowana. Celowanie w ATP c-Met przez drobnocząsteczkowe inhibitory jest jedną ze strategii hamowania kinazy tyrozynowej.
Historia

Na początku lat 80. MET opisano jako produkt białkowy transformującego onkogenu .


Wstępne próby zidentyfikowania konkurencyjnych względem ATP inhibitorów c-Met w 2002 roku doprowadziły do odkrycia K252a , inhibitora podobnego do staurosporyny , który blokuje c-Met. K252a była pierwszą strukturą, którą rozwiązano w kompleksie z niefosforylowaną domeną kinazy MET. Tworzy dwa wiązania wodorowe między zawiasem a podjednostką piralokarbazolu.
Później zaprojektowano serię bardziej selektywnych inhibitorów c-Met, w których rdzeń indolin-2-onu (zaznaczony kółkiem na rycinie 1) był obecny w kilku inhibitorach kinazy. SU-11274 wyewoluował przez podstawienie w pozycji 5 indolinonu i dodanie grupy 3,5-dimetylopirolu , powstał PHA-665752 – inhibitor drugiej generacji o lepszej sile i aktywności.
Zainteresowanie tą dziedziną gwałtownie wzrosło od 2007 roku, aw połowie 2009 roku opublikowano ponad 70 zgłoszeń patentowych.
przemyśle farmaceutycznym podjęto intensywne wysiłki po zaakceptowaniu c-Met jako odpowiedniego celu w terapii raka. Opublikowano 20 struktur krystalicznych z ligandami i bez ligandów , aw 2010 roku prawie tuzin małocząsteczkowych inhibitorów c-Met zostało przetestowanych klinicznie.
Wstęp
Receptorowe kinazy tyrozynowe (RTK) są istotnym elementem regulującym wiele wewnątrzkomórkowych szlaków transdukcji sygnału. Met kinaza tyrozynowa jest receptorem czynnika wzrostu hepatocytów (HGF), znanego również jako czynnik rozproszenia (SF). HGF ulega głównie ekspresji na komórkach nabłonka i komórkach mezenchymalnych , na przykład komórkach mięśni gładkich i fibroblastach . HGF jest zwykle aktywny w gojeniu ran, regeneracji wątroby , prawidłowym rozwoju zarodka i ssaków , morfogenezie narządów .
Dysregulacja c-Met może być spowodowana nadekspresją, amplifikacją genu, mutacją , zależną od liganda pętlą auto- lub parakrynną lub przedwczesną aktywacją RTK. Wszystkie te czynniki wpływają na przeżycie komórek, ich proliferację i ruchliwość. Prowadzą także do nowotworów i odporności na terapie, które mają na celu ich leczenie. Pacjenci z nieprawidłową aktywnością c-Met mają zwykle złe rokowanie , agresywną chorobę, zwiększoną liczbę przerzutów i krótszy czas przeżycia. Właśnie dlatego celowanie w szlak sygnałowy HGF/c-MET nie zostało podjęte w leczeniu raka, a kilka różnych podejść terapeutycznych jest testowanych klinicznie. Zastosowano różne podejścia do celowania w c-Met, z których każde skupiało się na jednym z kolejnych etapów, które regulują aktywację c-Met przez przeciwciała , agonistów peptydów , receptory wabików i inne inhibitory biologiczne lub inhibitory małocząsteczkowe.
Struktura i funkcja


Podrodzina c-Met RTK różni się strukturą od wielu innych rodzin RTK: dojrzała postać ma zewnątrzkomórkowy łańcuch α (50 kDa) i transbłonowy łańcuch β (140 kDa), które są połączone ze sobą wiązaniem dwusiarczkowym. Łańcuch beta zawiera wewnątrzkomórkową domenę kinazy tyrozynowej i ogon na C-końcu, który jest niezbędny do dokowania substratów i dalszej sygnalizacji.
HGF jest naturalnym ligandem o wysokim powinowactwie dla Met. Jego region N-końcowy wiąże się z Met, a dimeryzacja receptora i autofosforylacja dwóch tyrozyn zachodzą w pętli aktywacyjnej (pętla A) w domenie kinazowej Met.
Fosforylacja zachodzi w tyrozynach w pobliżu C-końca, tworząc wielofunkcyjne miejsce dokowania, które rekrutuje białka adaptorowe i prowadzi do dalszej sygnalizacji. Sygnalizacja odbywa się za pośrednictwem Ras/Mapk, PI3K/Akt, c-Src i STAT3/5 i obejmuje proliferację komórek, zmniejszoną apoptozę, zmienioną cytoszkieletu i inne.
Domena kinazy zwykle składa się z dwupłatowej struktury, w której płaty są połączone regionem zawiasowym, przylegającym do bardzo konserwatywnego miejsca wiązania ATP.
Rozwój
Korzystając z informacji uzyskanych ze struktury kokrystalicznej PHA-66752 i c-Met, zaprojektowano selektywny inhibitor PF-2341066. W 2010 roku przechodził badania kliniczne fazy I/II. Zamiana serii związków 4-fenoksychinolinowych na grupę acylotiomocznikową doprowadziła do powstania związków o aktywności c-Met, np. chinoliny . Był to kluczowy krok w rozwoju inhibitora c-Met, ponieważ wiązanie acylu daje końcowej grupie arylowej zdolność penetracji głębokiej kieszeni hydrofobowej , a tym samym zwiększa siłę działania związków. Znaleziono alternatywy dla wiązania acylotiomocznika, które mają pirymidonową , jak w AM7.
AM7 i SU11274 dostarczyły pierwszego dowodu, że można zidentyfikować stosunkowo selektywne inhibitory c-Met i że hamowanie prowadzi do efektu przeciwnowotworowego in vivo . Kiedy porównano struktury kokrystaliczne AM7 i SU11274 z c-Met, stwierdzono, że są różne: SU-11274 wiąże się w sąsiedztwie regionu zawiasowego z konformacją w kształcie litery U; ale AM7 wiąże się z c-Met w rozszerzonej konformacji, która obejmuje obszar od regionu zawiasowego do C-helisy. Następnie wiąże się w hydrofobowej kieszeni. c-Met przyjmuje nieaktywną, niefosforylowaną konformację z AM7, która może wiązać się zarówno z fosforylowaną, jak i niefosforylowaną konformacją kinazy.
Ze względu na te dwa różne rodzaje wiązania, małocząsteczkowe inhibitory Met zostały podzielone na dwie klasy; klasa I (podobna do SU-11274) i klasa II (podobna do AM7). Istnieje jednak inny rodzaj małocząsteczkowych inhibitorów, który nie pasuje do żadnej z tych dwóch klas; niekonkurencyjny inhibitor ATP, który wiąże się w inny sposób niż pozostałe dwa.
Inhibitory małocząsteczkowe różnią się selektywnością, są albo bardzo specyficzne, albo mają szeroką selektywność. Są albo konkurencyjne, albo niekonkurencyjne w ATP.
Małocząsteczkowe inhibitory c-Met konkurencyjne wobec ATP
Chociaż te dwie klasy różnią się strukturalnie, mają pewne wspólne właściwości: obie wiążą się w regionie zawiasowym kinazy (chociaż zajmują różne części miejsca aktywnego c-Met) i wszystkie mają na celu naśladowanie puryny ATP . BMS-777607 i PF-02341066 mają grupę 2-amino-pirydynową, AMG-458 ma grupę chinolinową , a MK-2461 ma tricykliczną grupę aromatyczną.
klasa I
Inhibitory klasy I mają wiele różnych struktur, są stosunkowo selektywne i mają konformację w kształcie litery U oraz wiążą się z pętlą aktywacyjną c-Met.
Związek struktura-aktywność inhibitorów klasy I

Odkryto serię triazolotriazyn, które okazały się bardzo obiecujące jako inhibitory c-MET. Zależność struktura-aktywność ( SAR) implikuje konieczność grupy arylowej połączonej z pierścieniem triazynowym i odpowiedniego akceptora wiązania wodorowego ( np. i że triazyna oddziałuje z Tyr1230. Znaleziono i zbadano szereg podobnych analogów. Strukturalnie podobne serie inhibitorów c-Met, w których fenolowy element wiążący zawias był połączony z aryloamino-triazolopirydazyną lub arylo-triazolotiapirydazyną. Łącznik jednoatomowy był bardziej wydajny niż łącznik dwuatomowy i wydawało się, że to podstawienie w pozycji benzylowej jest tolerowane. Opisano związki z heterocyklicznymi elementami wiążącymi zawias (chinolina, pirydyna , azaindol) połączonymi ze skondensowanymi, azotowymi heteroaromatykami (triazolopirydazyny, triazolopirazyny i triazolotriazyny). Patrz rysunek 4, aby uzyskać szczegółowe informacje.
Przykłady inhibitorów klasy I
JNJ-38877605, który zawiera łącznik difluorometylowy i biodostępną grupę chinolinową, przechodził w 2010 roku badania kliniczne fazy I dotyczące zaawansowanych i opornych na leczenie guzów litych. Badanie zakończono przedwcześnie z powodu toksyczności dla nerek spowodowanej przez metabolity środka.
PF-04217903, konkurencyjny wobec ATP i wyjątkowo selektywny związek, ma grupę N-hydroksyetylopirazolową związaną z C-7 triazolopirazyny. W 2010 roku przechodził I fazę badań klinicznych. [ wymaga aktualizacji ]
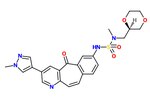
Zbadano SAR unikalnego rusztowania inhibitora kinazy o silnym działaniu hamującym c-Met, MK-2461. Azot pirydyny jest niezbędny do działania hamującego i zmniejszonej siły nasycenia centralnego pierścienia. Planarność cząsteczki okazała się niezbędna dla maksymalnej mocy. Cykliczne etery równoważą akceptowalną aktywność komórkową i farmakokinetyczne . Za kluczowe w procesie optymalizacji uważa się następujące elementy:
1) Grupy arylowe w pozycji 7, jak gdyby w celu zmaksymalizowania hydrofobowego upakowania i płaskości,
2) Ciasny SAR po dodaniu grupy sulfonamidowej i
3) Stosunkowo płaskie SAR grup narażonych na działanie rozpuszczalnika.
Często mutacje onkogenne c-Met powodują oporność na inhibitory drobnocząsteczkowe. W związku z tym analog MK-2461 był testowany przeciwko różnym mutantom c-Met, ale okazał się nie mniej skuteczny przeciwko nim. Daje to cząsteczce dużą przewagę w leczeniu nowotworów spowodowanych dysregulacją c-Met. MK-2461 był poddawany próbom eskalacji dawki I fazy w 2010 roku. [ wymaga aktualizacji ]
Klasa II
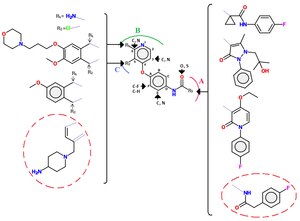
Inhibitory klasy II zwykle nie są tak selektywne jak inhibitory klasy I. Grupy mocznikowe są również wspólną cechą inhibitorów klasy II, zarówno w postaci cyklicznej, jak i acyklicznej. Klasa II inhibitorów zawiera wiele różnych cząsteczek, których wspólne rusztowanie można zobaczyć na rycinie 4.
Związek struktura-aktywność inhibitorów klasy II
Zbadano serię chinolinowych inhibitorów c-Met z wiązaniem acylotiomocznika. Znaleziono wiele serii analogów z alternatywnymi grupami wiążącymi zawiasy (np. zastąpienie grupy chinolinowej), zastąpienie wiązania tiomocznikowego ( np. malonamid, oksalamid, pirazolony) i ograniczenie fragmentu struktury acyklicznego acylotiomocznika różnymi heterocyklami aromatycznymi. Dalsze udoskonalenie obejmowało zablokowanie pozycji p wiszącego pierścienia fenylowego fluoru . Przykłady interakcji między c-Met a małymi cząsteczkami (zaznaczonymi czerwonym kółkiem) klasy II są następujące: Rusztowanie c-Met osadza się w kieszeni ATP trzema kluczowymi wiązaniami wodorowymi, końcowa amina oddziałuje z kieszenią rybozy (z ATP), końcowa grupa 4-fluorofenylowa jest zorientowana w hydrofobowej kieszeni, a pirolotriazyna pełni rolę grupy wiążącej zawias.
Przykłady inhibitorów klasy II
W badaniach klinicznych II fazy GSK 1363089 (XL880, foretynib) był dobrze tolerowany. Prowadziło to do niewielkich regresji lub stabilizacji choroby u pacjentów z rakiem brodawkowatym nerki i niskozróżnicowanym rakiem żołądka.
AMG 458 jest silnym małocząsteczkowym inhibitorem c-MET, który okazał się mieć ponad 100-krotną selektywność wobec c-MET w panelu 55 kinaz. Ponadto AMG 458 był w 100% biodostępny u różnych gatunków, a wewnętrzny okres półtrwania wydłużał się u wyższych ssaków.
Niekompetycyjne z ATP małocząsteczkowe inhibitory c-Met
Tiwantynib

Tiwantynib (ARQ197) jest selektywnym, biodostępnym po podaniu doustnym, zaawansowanym klinicznie niskocząsteczkowym i dobrze tolerowanym inhibitorem c-MET, który jest obecnie [ kiedy? ] w badaniach klinicznych fazy III u pacjentów z niedrobnokomórkowym rakiem płuca . ARQ197 jest niekonkurencyjnym względem ATP inhibitorem autofosforylacji c-MET o wysokiej selektywności względem niefosforylowanej konformacji kinazy. Tiwantynib przerywa interakcje między kluczowymi katalitycznymi . Struktura tiwantynibu w kompleksie z domeną kinazy c-Met pokazuje, że inhibitor wiąże konformację, która różni się od opublikowanych struktur kinazy. Tiwantynib silnie hamuje autoaktywację c-Met poprzez selektywne ukierunkowanie na nieaktywną postać kinazy pomiędzy płatami N i C i zajmuje miejsce wiązania ATP.
Badania kliniczne i zezwolenia regulacyjne
Stan na 2010 rok
Od czasu odkrycia Met i HGF wiele badań skupiło się na ich roli w raku. Szlak Met jest jednym z najczęściej rozregulowanych szlaków w raku człowieka. Lepsze zrozumienie trybów wiązania i projektu strukturalnego przybliża nas do wykorzystania innych interakcji białek i kieszeni wiążących, tworząc inhibitory o alternatywnych strukturach i zoptymalizowanych profilach.

Od 2010 roku w klinice badano kilkanaście inhibitorów szlaku Met, o różnych profilach selektywności kinazy, od wysoce selektywnych do wielokierunkowych, i osiągnięto duży postęp (patrz tabela 1). (np. XL184 (Kabozantynib), XL880 , ARQ197 ) [ wymaga aktualizacji ]
Zastosowanie inhibitorów c-Met z innymi środkami terapeutycznymi może mieć kluczowe znaczenie dla przezwyciężenia potencjalnej oporności, jak również dla poprawy ogólnej korzyści klinicznej. Inhibitory szlaku Met można stosować w połączeniu z innymi terapiami, w tym chemio- , radio- lub immunoterapią , jak również różnymi inhibitorami szlaku Met, np. z biologicznymi antagonistami HGF i Met lub przeciwciałami przeciwko HGF i MET. Nadal istnieje ryzyko kumulacji toksyczności i interakcji z innymi lekami.
Od 2010
W 2011 roku PF-02341066 (obecnie nazywany kryzotynibem) został zatwierdzony przez amerykańską FDA do leczenia niektórych niedrobnokomórkowych raków płuca .
W 2012 roku XL184/cabozantinib uzyskał aprobatę FDA do leczenia raka rdzeniastego tarczycy , aw 2016 roku uzyskał aprobatę FDA i UE do leczenia raka nerki.
Badania nad innymi inhibitorami
Tepotinib , (MSC 2156119J),
przedstawił wyniki badań klinicznych II fazy dotyczących raka płuc. We wrześniu 2019 roku Tepotinib otrzymał terapii przełomowej od Amerykańskiej Agencji ds. Żywności i Leków (FDA). W Japonii w listopadzie 2019 roku otrzymał oznaczenie leku sierocego , a we wrześniu 2020 roku w Australii.
Zobacz też
- Przejście mezenchymalne-nabłonkowe
- Czynnik wzrostu hepatocytów
- K252a
- Przejście nabłonkowo-mezenchymalne
- c-Met